Today, I review, link to, and excerpt from Nature‘s Obesogens: a unifying theory for the global rise in obesity [PubMed Abstract] [Full-Test HTML] [Full-Text PDF]. Int J Obes (Lond). 2024 Apr;48(4):449-460. doi: 10.1038/s41366-024-01460-3. Epub 2024 Jan 11.
There are 101 similar articles in PubMed.
The above article has been cited by 27 articles in PubMed.
All that follows is from the above article.
- Abstract
- Introduction and Background
- Ontogeny of obesity
- Current models of obesity
- The Energy Balance Model (EBM)
- The Carbohydrate-Insulin Model (CIM)
- The Energy Reduction-Oxidation Model (REDOX)
- The Obesogen Model (OBS)
- Proposed Integrated Model
- Summary
- Future directions
- References
- Funding
- Ethics declarations
- Additional information
- Rights and permissions
- About this article
Abstract
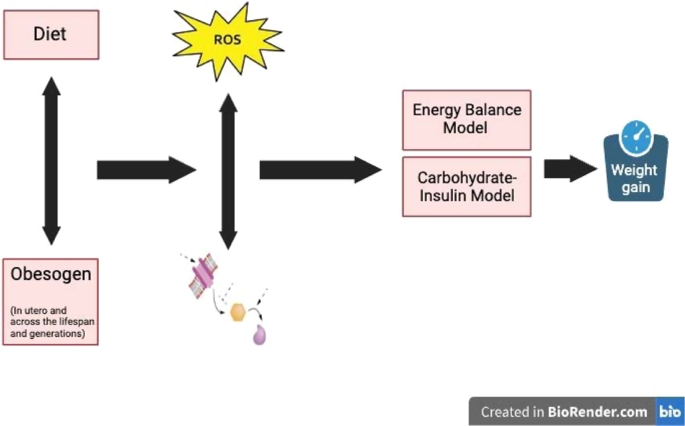
Despite varied treatment, mitigation, and prevention efforts, the global prevalence and severity of obesity continue to worsen. Here we propose a combined model of obesity, a unifying paradigm that links four general models: the energy balance model (EBM), based on calories as the driver of weight gain; the carbohydrate-insulin model (CIM), based on insulin as a driver of energy storage; the oxidation-reduction model (REDOX), based on reactive oxygen species (ROS) as a driver of altered metabolic signaling; and the obesogens model (OBS), which proposes that environmental chemicals interfere with hormonal signaling leading to adiposity. We propose a combined OBS/REDOX model in which environmental chemicals (in air, food, food packaging, and household products) generate false autocrine and endocrine metabolic signals, including ROS, that subvert standard regulatory energy mechanisms, increase basal and stimulated insulin secretion, disrupt energy efficiency, and influence appetite and energy expenditure leading to weight gain. This combined model incorporates the data supporting the EBM and CIM models, thus creating one integrated model that covers significant aspects of all the mechanisms potentially contributing to the obesity pandemic. Importantly, the OBS/REDOX model provides a rationale and approach for future preventative efforts based on environmental chemical exposure reduction.
Introduction and Background
Obesity continues to increase at an alarming rate across the globe despite an increase in the number of diets and drugs [1]. The etiology of obesity is still not understood, as evidenced by the following statements from recent articles:
- 1.In 2017, an Endocrine Society Scientific Statement [2] noted, “The current lack of consensus regarding obesity pathogenesis has resulted in competing and poorly justified claims both from within and outside the scientific community. These inconsistencies erode public trust and confidence in the scientific process concerning obesity and its treatment, further supporting nonscientific ideologies and products.”
- 2.A recent perspective noted that we do not have a clear explanation for the obesity epidemic [3]. Notably, the national data do not support higher energy consumption as a driver of the obesity epidemic since 2000. “This lack of adequate attention and investment in understanding the root causes of the obesity epidemic … may at least partly owe to the belief that the foundational causes are already known” [3].
- 3.A recent scientific meeting organized at the Royal Society in London by Profs. Speakman, Sørensen, Hall, and Allison focused on “Causes of obesity: theories, conjectures and evidence” [4]. Despite numerous symposia, guidelines, and punditry, the attendees were no closer to a unifying theory for the global rise in obesity.
Ontogeny of obesity
Obesity is a neuroendocrine disease [2]. Body weight is highly regulated by various systems and hormones from many tissues integrated by the brain to regulate food intake and metabolism [5]. Key questions include, what has changed over the last 50 years that led to the obesity epidemic? What has been imposed on or removed from society that led to the obesity epidemic?
Before examining the various models of obesity, it is essential to understand when obesity starts (ontogeny), as that aspect of etiology must be integrated into any model. Obesity, like other non-communicable diseases, can have at least some of its origins in utero and early childhood and may manifest itself at any time across the lifespan. Both under and over-nutrition in utero are associated with obesity in the offspring later in life [6,7,8,9,10]. Mothers or fathers who are overweight during pregnancy may have overweight offspring [11]. In a rodent study, maternal exercise during pregnancy promoted physical activity in adult offspring, suggesting that the propensity to exercise may also be programmed during development [12]. The strongest perinatal predictor of childhood obesity is reported to be maternal pre-pregnancy obesity [13]. In a rodent study, gestational exposure to a Western diet predisposes to a high fat and sugar diet in later life to promulgate obesity [14]. Developmental programming can also affect intergenerational obesity in humans [15, 16], and transgenerational epigenetic inheritance of obesity is seen in animal models [17]. Altered epigenetic regulation of gene expression during development due to nutrition, stress, or environmental chemicals can interfere with the control of food intake and metabolism, including metabolic efficiency via effects on the development of the adipose tissue, pancreas, liver, gastrointestinal tract, brain and/or muscle, thereby resulting in an altered body weight set point or sensitivity for developing obesity across the lifespan and generations [18]. In utero and early development may be a highly sensitive time for the programming of fat storage due to permanent effects on gene expression and adipose tissue differentiation. Consequently, nutrition, stress, and environmental chemicals can all have the potential to alter metabolic signaling at this stage, leading to excessive adipose tissue growth and energy storage throughout life.
Current models of obesity
Two of the major models of obesity include the energy balance model (EBM) reviewed in [5, 19], which emphasizes overeating and sedentary activity, and the carbohydrate-insulin model (CIM) reviewed in [20], which emphasizes energy storage due to hyperinsulinemia’s effect on adipocytes. The reduction-oxidation model (REDOX) is an additional lesser-known model reviewed in [21, 22]. The REDOX model emphasizes that many substances, including processed foods and environmental exposures, can cause obesity by generating false and misleading information about energy status. This misinformation is driven by changes in the oxidation-reduction potential of metabolites that circulate and communicate to organs throughout the body. A fourth model, the obesogen model (OBS) reviewed in [18], posits that exposure to environmental chemicals, especially during critical developmental periods, but also across the lifespan, can affect long-term metabolism via hormonal changes, increasing susceptibility to obesity.
Here we discuss these four models in more detail. Each model focuses on a specific aspect of obesity: neural control and calories (EBM); carbohydrates and insulin (CIM); metabolic oxidation-reduction mismatches (REDOX); and developmental exposures to environmental stimuli (OBS). Each model is usually presented as an exclusive and non-overlapping archetype responsible for the increase in obesity; however, below, we propose a more integrated approach. We describe, in turn, each model, the integration of the OBS and REDOX models, and finally propose that this OBS/REDOX model can account for much of the data that support both the EBM and CIM models.
The Energy Balance Model (EBM)
According to the EBM, obesity is a disorder of energy balance. Overweight and obesity result from a chronic imbalance between energy intake and expenditure (21, 25, 26); we gain weight because we eat more, burn fewer calories, or both. The EBM proposes that the brain is the primary organ responsible for body weight regulation via the integration of internal and external signals by mediators not yet defined and that disruption of normal signals leads to overeating and obesity. In this model, it is food intake that needs to be controlled. The EBM notes that consuming ultra-processed food (UPF) causes overeating, increasing adiposity, insulin resistance, consequent insulin compensatory secretion, and resultant weight gain [23]. Recent additions to the EBM include other gastrointestinal hormones (e.g., glucagon-like peptide 1 (GLP-1), peptide YY3-36 (PYY), and gastric inhibitory polypeptide (GIP)), all of which reduce acute food intake [24]. GLP-1 acts centrally [25] and peripherally [26] to inhibit food intake. Indeed, newer GLP-1 analogs have become primary therapies for T2D and obesity [27]. However, it should also be noted that GLP-1 analogs may also have untoward side-effects by delaying gastric emptying, leading to nausea, vomiting, and gastroparesis [28]. These side-effects may be part of the mechanism for the weight reduction, as demonstrated by the loss of equal amounts of muscle and fat, consistent with anorexia and/or starvation [29].
The gut microbiome may also support the EBM to predispose to obesity. Animal studies argue that changes in the microbiome increase energy availability by increasing energy harvest efficiency [30]. Several investigators have demonstrated changes in the human microbiome, paralleling changes in the diet [31,32,33] suggesting that one mechanism of diet-induced obesity may be through microbiome-promotion of altered energy harvesting [34]. Animal models have also provided evidence that a “predisposed” microbiome might increase both energy intake (via central mechanisms) and energy absorption (via gastrointestinal mechanisms) to contribute to obesity [35]. However, while changes in diet (and therefore by inference obesogens) have effects on the gut microbiota, there are currently no compelling data thus far that differentiates between consequence and cause. Furthermore, human data addressing this mechanism have been somewhat inconsistent [36]. “Randomized controlled trials of microbiota transfer in human participants have not shown effects on body weight. With a more critical reading, early studies did not show as large an effect as first appeared and later research, including human trials, has failed to support a role of the gut microbiota in shaping body weight” [37].
The EBM proposes that achieving a stable weight is as simple as balancing energy intake versus expenditure; however, a recent review [38] concluded that weight stability is much more complex. Most individuals’ experimentally induced weight gain or loss has no lasting effects. The original weight is rapidly re-established when the controlled feeding experiment ends [39]. In addition, these studies documented that many more calories than predicted were needed to gain weight. Conversely, a much more significant caloric decrease was required than expected to lose weight, indicating a strong biochemical regulatory mechanism for weight maintenance [40].
Exercise has never been shown to strongly modulate body weight, possibly due to compensatory regulation of energy efficiency and the repartitioning of fat into muscle, likely due to growth hormone secretion [41]. It should be noted that the amount of energy expenditure caused by increased physical activity does not translate directly to weight loss, since if it did, people would lose more weight than they do in trials in which physical activity is increased under close supervision [42].
The EBM does not explain why numerous animal species (both in the wild, near human populations, and in captivity with controlled diets) have all gained weight over the past 25 years [43]. In addition, since 2000, obesity rates have increased while energy intake decreased and energy expenditure increased [3]. The EBM also does not explain why, for a given caloric intake or physical activity, BMI was higher in 2006 than in 1988 [44]. Lastly, the EBM does not address how diet or environmental exposures during development influence later-life obesity.
The Carbohydrate-Insulin Model (CIM)
The obesity epidemic in the U.S. temporally coincided with the food industry and the federal government’s promotion in the 1970s of low-fat diets and the resulting increased intake of refined carbohydrates and fructose-containing sweeteners, reviewed in [45]. This change was based on the epidemiologic correlation of dietary fat, low-density lipoprotein, cholesterol, and cardiovascular mortality [46]. However, the inevitable result of this paradigm change was increased carbohydrate consumption with induced insulin response, increased energy deposition into adipose tissue, with increased obesity and related chronic non-communicable diseases.
The CIM posits that a diet high in rapidly digestible carbohydrates causes an elevated insulin response that stimulates lipoprotein lipase (LPL) and suppresses the adrenergic system and lipolysis in adipose tissue, thus promoting lipogenesis [20, 47]. Therefore, the crux of the differences between the CIM and the EBM is two-fold. First, the CIM focuses on the endocrine response to the sources of dietary substrate, while the EBM focuses on the caloric content of the diet [20]. Second, the CIM focuses on fuel partitioning in the periphery (particularly adipose tissue), while the EBM focuses on the brain and its regulation of nutrient intake. However, the precise mechanisms still need to be resolved [19]. Both EBM and CIM stress the importance of diet; however, the EBM focuses on the quantity of calories, while CIM focuses on the quality of calories, specifically carbohydrates with a high glycemic index (GI) (i.e., higher insulin-stimulated response to carbohydrates). Carbohydrates produce higher insulin secretion levels, down-regulating the insulin receptor and leading to insulin resistance and altered signaling in the brain [48, 49]. On the other hand, restriction of such carbohydrates would result in lower insulin levels, reduced fat storage and increased lipolysis, and resultant weight loss. Indeed, increasing insulin promotes weight accrual in humans [50]; as demonstrated by type 1 diabetes (deficient insulin production); one of the cardinal symptoms of the disorder is weight (especially adipose tissue) loss, while insulin supplementation rapidly increases weight gain and adiposity. Pima Indians who have high rates of obesity also have significantly higher fasting insulin and display a higher amplitude insulin response to a glucose load [51].
UPF in the Western diet may also induce nutritional insufficiencies detrimental to the brain, resulting in a lack of critical nutrients vital for neurotransmitter function, cognition, mood, sleep and optimal neurodevelopment [52, 53]. Thus, the Western UPF-rich diet may play an essential role in the CIM model of obesity, as it does for the EBM model.
The Energy Reduction-Oxidation Model (REDOX)
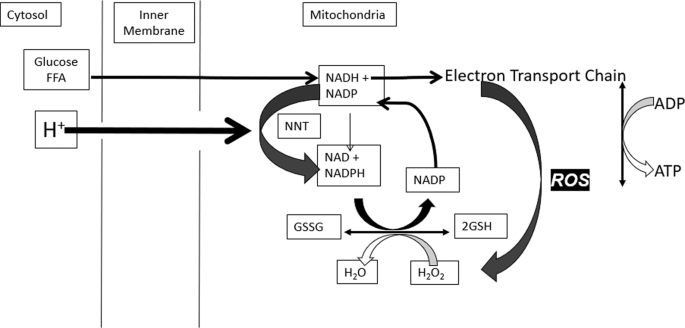
Mitochondria are the primary site of cellular ATP or energy production. Reactive oxygen species (ROS) is a natural signal that all mitochondria generate when energy needs have been met and fuel is still available. The value of this signal is to promote fuel storage via stimulation of insulin secretion. The short-lived signal stops when fuel has been stored and is no longer excessive. The mechanisms involved are biochemically complex: metabolism of glucose and fat generates reduced nicotinamide adenine dinucleotide (NADH) in the mitochondria. This fuel-derived NADH donates electrons to the electron transport chain (ETC) to maintain a high energy state or ATP/ADP ratio. When fuel supply exceeds the need for ATP production, elevated NADH produces ROS, an essential intracellular signal of fuel excess [21]. Excess fuel increases NADH and ROS rapidly in all cells, while lacking fuel decreases both. Rapid mitochondrial ROS removal requires NADPH, derived from glucose and fat metabolism. Redox reactants, therefore, comprise an energy-responsive communication system within each cell and cellular compartment [21, 54,55,56] (Fig. 1).
Although it is well-established that high levels of ROS can cause lipid peroxidation, protein denaturation, and cellular damage, the small increases in ROS that occur in response to acute fuel excess (assuming ATP adequacy) have significant positive cell-specific effects in metabolically active cells. ROS stimulates fat storage in adipocytes by regulating lipogenesis and lipolysis; the balance depends on the adipocyte’s hormonal milieu. For instance, elevated insulin stimulates LPL and inhibits lipolysis to drive TG synthesis, whereas catecholamines stimulate lipolysis. The hormonal milieu and ROS excess drive a net increase in TG stores in the presence of hyperinsulinemia or a net decrease when basal insulin is low [55]. In pancreatic ß-cells, ROS application externally or ROS generation internally stimulates insulin secretion [21, 57,58,59]. In the liver, a physiological increase in ROS reduces glucose output, whether of mitochondrial, cytosolic, or extracellular origin [55, 56]. ROS also acts in the hypothalamus to decrease food intake through effects on various neurons, including activation of pro-opiomelanocortin (POMC) neurons and suppression of agouti-related protein (AgRP)/neuropeptide Y (NPY) neurons [60]. Various hormones and nutrients also influence hypothalamic ROS generation [61]. Thus, the consequences of ROS production in response to excess glucose or fat supply are logical, synchronous, and coordinated: ß-cells release insulin to promote energy storage, adipocytes store triglycerides, hepatocytes stop gluconeogenesis, and neurons signal satiety.
Rapid ROS removal is achieved through catalase, glutathione, and thioredoxin systems. However, excess ROS production can exceed the capacity of thiol removal systems, resulting in oxidative damage to susceptible proteins and lipids, with resultant cell damage or death. The possibility that inadequate ROS removal systems could differentiate sensitive individuals from individuals that maintain average weight on similar diets has yet to be investigated.
People with obesity [62] and those who consume large quantities of UPF [63] appear to have lower antioxidant capacity, including superoxide dismutase, glutathione peroxidase, and catalase, compared to people of normal weight [64,65,66]. If this results from insufficient antioxidant capacity, it would be expected to increase oxidative stress. Normal energy-dependent mitochondrial ROS signals are transient, and ROS removal requires NADPH produced from NADH via nicotinamide nucleotide transhydrogenase (NNT) (Fig. 2). Since flux through NNT is driven by the proton gradient, it decreases the mitochondrial membrane potential, ultimately stimulating restoration by the ETC [67,68,69]. This “proton leak” is readily determined by measuring oxygen consumption when ATP production is inhibited by oligomycin [68, 69].
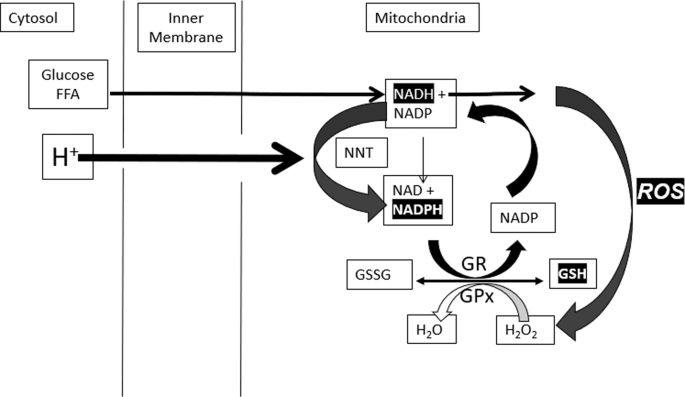
Fig. 2: Coordinated oscillations GSH due to ROS production and removal.
ROS induces oscillations in GSH that convert ROS to H2O (via GPX) NADPH to restore GSSG to GSH (via GR), NADH to restore NADPH (via NNT), membrane potential (via proton-driven NNT) and O2 (via ETC) to restore the proton gradient.
Since excess nutrient consumption, the consumption of UPF and exposure to obesogens can all lead to an increase in ROS, the timing of the increase in ROS corresponds to the increase in the rate of obesity which correlates to excess nutrient consumption, UPF and obesogens.
Additional support for the REDOX model comes from data showing that enlarged adipocytes in obesity are associated with chronic low-grade inflammation in adipose tissue and increased oxidative stress [70]. Adipose tissue induces the synthesis of pro-inflammatory cytokines (TNF-α, interleukins IL-1 and IL-6), thereby promoting additional ROS generation by macrophages and monocytes [70]. Excessive fat accumulation in people with obesity leads to increased circulating free fatty acid levels, which promote higher fat oxidation and increased ROS [71].
It is also essential to be aware that ROS impacts thousands of proteins containing susceptible sulfhydryl groups on cysteine residues [72]. These proteins, in turn, regulate cellular signal transduction, including endocrine secretion and energy homeostasis [73].
Weight maintenance has been well-documented to involve variations in energy efficiency [39]. We suggest that the “leak” resulting from flux through the mitochondrial NNT is a positive and critical element in maintaining stable body fuel stores and regulating energy efficiency [67, 74]. Thus, excess fuel generates ROS as a signal. Subsequently, ROS removal stimulates flux through NNT and “wastes” energy when fuel is plentiful but not when resources are scarce or mitochondrial membrane potential is low. ROS removal also depends on thiol availability and rapid restoration of reduced thiol. We propose that overwhelming this ROS handling mechanism to compensate for caloric variation will diminish energy-wasting capability, sustain elevated ROS levels, and promote oxidative damage and metabolic dysregulation.
Another possible mechanism of REDOX metabolites influencing obesity is through epigenetic changes [75]. Alterations in the human epigenome are seen during nutritional privation or deprivation alterations, especially during the fetal or neonatal period [76]. Increased ROS formation has been demonstrated to increase methylation status in some tissues [77] and ROS may alter adipose tissue differentiation during development [78]. From a mechanistic standpoint, weight gain can be mitigated by increasing the amount of dietary folic acid, which reduces ROS formation [79] and gene methylation [80].
Excess fuel stimulates ROS, leading to insulin secretion (CIM model), promoting fat storage and altering appetite (EBM model). The link to obesogens is based on the observation that obesogens cause oxidative stress which is a consequence of excess ROS or the failure to remove ROS adequately. Thus, we hypothesize that excess fuel alone or combined with obesogens generates toxic amounts of ROS that cause damage—changes in either pyridine nucleotides or ROS impact redox. Our model hypothesizes that such linked changes in ROS and redox, provide a common mechanism by which each model leads to obesity.
The Obesogen Model (OBS)
Obesogens are ingested or internalized chemicals that alter energy metabolism, increasing adiposity. Many act via alterations in endocrine signaling. They disrupt signaling pathways (e.g., hormone receptors, transcription factors, ROS) in various cell types and tissues that regulate energy intake and expenditure, nutrient handling, and adiposity. Indeed, they have been shown to act during development in animal models to disrupt adipose tissue development via increases in number, size, location, and function. They also alter the control of food intake and metabolic rate via effects on the pancreas, adipose tissue, liver, GI tract, brain and/or muscle, thereby altering the programming of the setpoint or sensitivity for developing obesity later in life [18].
As noted above, obesity can start in utero due to altered nutrition, the Developmental Origins of Health and Disease [81]. This same paradigm holds for obesogens: development is the most sensitive time for obesogen exposures to alter the epigenetic programming of developing metabolic tissues leading to tissues that “look” normal but have altered epigenetic profiles, leading to increased sensitivity to weight gain later in life [18, 82, 83]. Some characteristics of obesogen action during development include that subtle epigenetic changes may be detectable at birth but their effects may not be apparent until later in life e.g., a latency between exposure and weight gain which may last from months to decades, effects will likely be sex specific, the effects of developmental exposure to obesogens may not be apparent without a challenge or “second hit” later in life [18]. Thus, many studies of obesogen action focus on developmental exposure and effects on weight gain later in life. Obesogens can also act throughout the lifespan, where in most cases the effects may not be permanent, and across generations; transgenerational epigenetic inheritance [84, 85].
Obesogens can be natural (e.g., metals, viruses), anthropogenic prescription drugs, environmental (insecticides, plastics, household chemicals, particulate matter), or food components (fructose, trans-fats, preservatives, emulsifiers) [18, 86]. Obesogens include solvents (polychlorinated biphenyls (PCBs)); pesticides (e.g., dichlorodiphenyltrichloroethane (DDT), chlorpyrifos, diazinon, permethrin, neonicotinoids); non-stick coatings (e.g., per- and polyfluorinated substances (PFAS)); clothing and furniture protectants (e.g., polybrominated diphenyl ethers (PBDEs), organophosphate flame retardants (OPFRs)); food preservatives/additives/emulsifiers (e.g., parabens, monosodium glutamate, carboxymethylcellulose, 3-tert-butyl-4-hydroxyanisole (3-BHA)); personal care products (e.g., phthalates, parabens); plastics (e.g., phthalates, bisphenols); resins and can linings (e.g., bisphenols); and air pollutants (e.g., polycyclic aromatic hydrocarbons (PAHs), fine particulate matter (PM2.5)) [87]. Some pharmaceutical drugs [88, 89] and early-life antibiotics can also be obesogens. Exposures can occur via air, water, food, skin contact or dust inhalation [90, 91].
Obesogens include environmental chemicals that have arisen in the past 50–70 years with the first increase in the 1960s [92, 93], prior to the start of the increase in obesity in adults in the U.S. in the 1970s, and children a decade later as noted by NHANES studies [94, 95]. Everyone is now exposed to a variety of obesogenic chemicals. Human studies show that obesogens affect weight gain in various countries including Spain, Poland, Mexico, Denmark, Belgium, Greece among others indicating the global nature of the relation of obesogens to obesity [96,97,98]. Obesogens permeate our food supply (Fig. 3) and are often consumed unintentionally. They are also in our water supply and in the air we breathe.
Fig. 3: The Western ultra-processed food diet is obesogenic.
The Western Diet (left panel) per se is obesogenic. In addition (right panel), chemicals in food packaging, such as can linings, can contain obesogens (red) which can leach into the food. Many food additives, preservatives, emulsifiers, and antioxidants are obesogens. Many fruits and vegetables are sprayed with pesticides, and some residues remain on them. Potential obesogens are those with only in vitro data. Reviewed in [87, 146, 147].
Thousands of new chemicals have entered our food supply and environment since the obesity pandemic began. Hundreds of in vitro, animal, and human epidemiologic birth cohort studies show effects or associations between environmental chemicals and obesity, including systematic reviews and meta-analyses reviewed in [18, 96, 98,99,100]. While exposure is ubiquitous, the effects of obesogens vary depending on genetic susceptibility, age, sex, home and work location, personal habits, race, and diet. For example, African-Americans tend to be exposed to higher air pollutants, bisphenol A (BPA), phthalates, organochlorine pesticides, and PCBs because of their neighborhood environment, personal care products, and/or diet [101, 102].
Obesogens affect numerous metabolic endpoints across the lifespan, including adipocyte differentiation, adipocyte number, size, and function, lipid levels, the gut microbiome, food intake, energy expenditure, inflammation, and insulin resistance [18]. Similarly, obesogens can impact animals that share our environment [103,104,105]; perhaps obesogens can explain why even animals in captivity with controlled diets have gained weight over the last 25 years [43].
In September 2022, Healthy Environment and Endocrine Disruptor Strategies (HEEDS) held a workshop in Racine, WI to integrate the obesogen model into the thinking of mainstream basic, clinical obesity, and nutrition researchers [106]. A report from that meeting noted, “Based on the robust nature of the in vitro and animal model data on obesogens, the obesogen hypothesis/model of obesity should receive greater attention by the broader scientific community as a potential contributor to the obesity pandemic.” Fig. 4 overviews the OBS model. This workshop also outlined data gaps and needs for the OBS field. These include human data that show decreased obesogen exposure can improve metabolic health, leveraging clinical studies to establish causality, more experiments to understand the mechanism of obesogen action on the brain satiety and appetite centers and the hedonic, emotional eating center, and methods to determine the risk of obesity attributable to obesogens compared to diet, genetics and other factors.
Fig. 4: Integrating obesogen actions.