In addition to today’s resource, please see and review Embedding The Online Supplementary Material From “The Carbohydrate-Insulin Model
Posted on May 25, 2025 by Tom Wade MD.
Today, I review, link to, and excerpt from The carbohydrate-insulin model: a physiological perspective on the obesity pandemic [PubMed Abstract] [Full-Text HTML] [Full-Text PDF]. Am J Clin Nutr. 2021 Dec 1;114(6):1873-1885. doi: 10.1093/ajcn/nqab270.
There are 101 similar articles in PubMed.
The above article has been cited by 99 articles in PubMed.
All that follows is from the above article.
Abstract
According to a commonly held view, the obesity pandemic is caused by overconsumption of modern, highly palatable, energy-dense processed foods, exacerbated by a sedentary lifestyle. However, obesity rates remain at historic highs, despite a persistent focus on eating less and moving more, as guided by the energy balance model (EBM). This public health failure may arise from a fundamental limitation of the EBM itself. Conceptualizing obesity as a disorder of energy balance restates a principle of physics without considering the biological mechanisms that promote weight gain. An alternative paradigm, the carbohydrate-insulin model (CIM), proposes a reversal of causal direction. According to the CIM, increasing fat deposition in the body-resulting from the hormonal responses to a high-glycemic-load diet-drives positive energy balance. The CIM provides a conceptual framework with testable hypotheses for how various modifiable factors influence energy balance and fat storage. Rigorous research is needed to compare the validity of these 2 models, which have substantially different implications for obesity management, and to generate new models that best encompass the evidence.
Keywords: dietary carbohydrate; endocrinology; energy balance; glucagon; incretins; insulin; macronutrients; obesity; scholarly discourse; weight loss.
© The Author(s) 2021. Published by Oxford University Press on behalf of the American Society for Nutrition.
Introduction
The last century has witnessed fundamental developments in our understanding of the biological basis of obesity. Many lines of investigation demonstrate that body weight is controlled by complex and interconnected systems involving multiple organs, hormones, and metabolic pathways. Common genetic variants, acting on these systems, may explain >20% of the population-level variation in BMI (1). Rare mutations have been identified that cause obesity in humans (2). Animal models have been generated in which mutation of a specific gene yields susceptibility to, or protection from, obesity (3). These genetic and molecular insights lay the foundation for many scientific models of obesity, helping to explain individual susceptibility to changing environmental conditions. However, in view of the rapid worldwide increase in BMI despite relatively stable genetic susceptibility, scientific explanations for the pandemic must consider environmental factors.
During the last century, 2 models addressing environmental causes of obesity have emerged. In the dominant energy balance model (EBM), energy-dense, tasty, modern processed foods drive a positive energy balance through increased intake, and thereby result in fat deposition. In the carbohydrate-insulin model (CIM), a crucial effect of diet is metabolic, by influencing substrate partitioning. Rapidly digestible carbohydrates, acting through insulin and other hormones, cause increased fat deposition, and thereby drive a positive energy balance.
In this review, we provide the most comprehensive formulation of the CIM to date, argue that the CIM better reflects knowledge on the biology of body weight control than the EBM, specify testable hypotheses to help resolve controversies, and call for constructive discourse among scientific camps on this question of critical public health importance.
Obesity Conceptualized as an Energy Balance Disorder
Obesity is commonly considered a disorder of energy balance. Surrounded by highly palatable, heavily marketed processed foods, many people consume more energy than they burn—an imbalance exacerbated by sedentary lifestyles—with the surplus deposited into body fat. According to this view, the foundation of obesity management is control of energy balance by decreasing intake (“eat less”) and increasing expenditure (“move more”) (Supplemental Table 1). For instance, the USDA Dietary Guidelines for Americans 2020–2025 state that, “Losing weight … requires adults to reduce the number of calories they get from foods and beverages and increase the amount expended through physical activity” (4). A 2013 expert report from several major professional health associations asserts, “To achieve weight loss, an energy deficit is required” [High evidence strength] (5). An influential textbook concludes, “All diets that result in weight loss do so on one basis and one basis only: they reduce total calorie intake” (6).
Importantly, the EBM considers all calories metabolically alike for practical purposes (7). Thus, specific foods or diets may produce variable amounts of weight gain or loss, but virtually only through energy intake related to hunger, satiation, satiety, hedonic effects, or “passive overconsumption” (8). Dietary treatment focuses on behavioral strategies to help people reduce energy intake, especially of the energy-dense, tasty, processed foods thought to drive a positive energy balance, and to manage adverse effects (e.g., hunger).
Tautologies and Other Limitations of the EBM
Energy balance and body weight
The appealing simplicity of the EBM belies an inherent tautology. Weight gain (or more precisely, fat gain) can occur only with a positive energy balance, in the same way that a fever can occur only if the body generates more heat than it dissipates. However, these reiterations of the law of energy conservation ignore causality. During the pubertal growth spurt, energy intake exceeds expenditure as body energy stores increase. Does increased consumption drive growth or does growth drive increased consumption? Neither possibility violates laws of physics, but the 2 perspectives have fundamentally different physiological bases and implications. Because the relation between energy balance and weight change is inseverable, statements regarding the importance of a negative balance provide no meaningful information about etiology.
Palatability and food intake
Regarding dietary drivers of obesity, common versions of the EBM focus on the variety and availability of “hyper-palatable” (9), energy-dense, processed foods (Supplemental Table 1). Clearly, people tend to eat more of the foods that they find tasty, and palatability seems to influence short-term food selection and energy intake. However, surprisingly little evidence relates palatability directly to chronic overconsumption (i.e., relative to energy requirements) in laboratory animals or humans under normal conditions (10–12).
Compared to a standard diet, a cafeteria diet composed of a variety of presumably palatable human “junk foods” causes rodents to gain weight (13). Importantly, this diet differs in macronutrients, sugar, saturated fat, and fiber; differences in composition, not palatability, appear to cause the weight gain (10, 14, 15). To examine this issue, Tordoff et al. (10) added highly palatable nonnutritive flavors to a standard rodent diet. When offered a choice, mice showed a strong, persistent preference for the flavored over the unflavored diet. However, the flavored diet increased neither food intake nor weight gain. In a complementary design, Ramirez (15) reported that rats rejected a bitter liquid diet in favor of a plain solid diet of the same nutrient composition when given a choice, but that rats given only the bitter liquid diet increased energy intake and weight compared with those given only the solid diet.
As reviewed by Johnson and Wardle (11), no interventional studies in humans have demonstrated a causal relation between palatability and obesity, controlling for confounding factors such as macronutrient composition. Moreover, major putative contributors to palatability—dietary fat (16), energy density (17), food variety (18), and food processing per se (19, 20)—have not been shown to cause long-term weight gain.
The very notion of palatability seems to lack an operational definition beyond “fast foods,” foods high in “fat, sugar, and salt,” or “ultra-processed” foods (9). In fact, palatability is not a fixed property of food, but rather modifiable through learning and influenced by physiological state. In humans, insulin administration associated with mild hypoglycemia preferentially activates limbic-striatal brain regions, promoting a greater desire for high-calorie foods in general and possibly high-carbohydrate foods (21–23). In the absence of clear correlates to intrinsic food properties, hyper-palatable foods have been defined as those that drive food intake—another tautology of the EBM, which simultaneously attributes increased food intake to hyper-palatable foods.
Anomalies
By focusing on energy balance—characteristically through conscious control, as highlighted in Supplemental Table 1—the basic formulation of the EBM essentially disregards knowledge about the biological influences on fat storage (24, 25). Moreover, a central conundrum is to understand why the so-called body weight “set point” (7) has increased rapidly among genetically stable populations. In the 1960s, the average man in the United States weighed ∼75 kg. Providing excess dietary energy to increase his weight to 90 kg would have elicited biological responses (e.g., decreased hunger, increased energy expenditure) to resist that gain. Today, the average man weighs ∼90 kg; restricting energy to reduce his weight to 75 kg would elicit opposite responses (26–30). By excluding a metabolic effect of diet, common versions of the EBM offer no explanation for what changes in the environment have dysregulated the biological systems that counteract energy imbalance and resist weight change.
Furthermore, conceptual adoption of the EBM has failed to stem the obesity pandemic. Governments and professional health organizations heavily promote energy restriction (especially with low-fat diets) and exercise; nutrition labels on packaged foods prominently display calorie content; and personal responsibility to avoid excess weight gain is emphasized in patient care. Nevertheless, obesity prevalence continues to increase worldwide, prompting spectacularly complex formulations of the EBM addressing myriad biological, behavioral, environmental, and societal factors converging on energy balance (Supplemental Figure 1), with questionable practical translation.
The CIM as a Physiological Explanation for the Obesity Pandemic
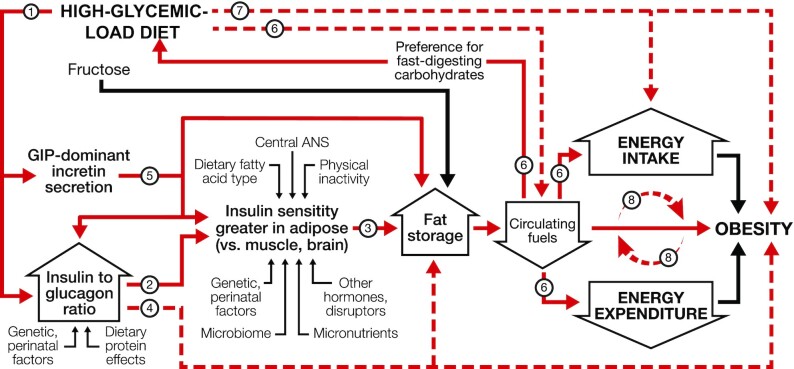
Like the EBM, the CIM posits that changes in food quality drive weight gain. However, according to the CIM, hormonal and metabolic responses to the source of dietary calories, not merely calorie content, lie upstream in the mechanistic pathway. In other words, the CIM proposes a reversal of causal direction: over the long term, a positive energy balance does not cause increasing adiposity; rather, a shift in substrate partitioning favoring fat storage drives a positive energy balance, as shown in Figure 1. Among modifiable factors, dietary glycemic load (GL) has central importance.
Dynamic phase of obesity development in the carbohydrate-insulin model. The relation of energy intake and expenditure to obesity is congruent with the conventional model. However, these components of energy balance are proximate, not root, causes of weight gain. In the compensatory phase (not depicted), insulin resistance increases, and weight gain slows, as circulating fuel concentration rises. (Circulating fuels, as measured in blood, are a proxy for fuel sensing and substrate oxidation in key organs.) Other hormones with effects on adipocytes include sex steroids and cortisol. Fructose may promote hepatic de novo lipogenesis and affect intestinal function, among other actions, through mechanisms independent of, and synergistic with, glucose. Solid red arrows indicate sequential steps in the central causal pathway; associated numbers indicate testable hypotheses as considered in the text. Interrupted red arrows and associated numbers indicate testable hypotheses comprising multiple causal steps. Black arrows indicate other relations. ANS, autonomic nervous system; GIP, glucose-dependent insulinotropic peptide.
GL* is defined as the arithmetic product of total carbohydrate amount and glycemic index (GI—a measure of how foods with controlled amounts of carbohydrate raise postprandial blood glucose) (31). GL is a validated predictor of postprandial glycemia with consumption of typical foods, meals, or eating patterns (32–34). High-GL foods include processed grains, potato products, and foods with a high free sugar content (34). Most fresh whole fruits, minimally processed grains, legumes, nuts, and nonstarchy vegetables have moderate or low GL. Fats and proteins, with no direct impact on postprandial blood glucose, have a nominal GL of 0.
*Glycemic Load (GL): This refers to the effect of a food on blood sugar levels, taking into account both the glycemic index (GI) and the amount of carbohydrates in a serving. A food with a high GL will cause a greater rise in blood sugar than a food with a low GL, even if they have the same GI.
The rapid absorption of glucose after consumption of a high-GL meal increases insulin secretion, suppresses glucagon secretion, and elicits a glucose-dependent insulinotropic polypeptide (GIP)-dominant incretin response (35–37). This highly anabolic state, for the first hours after eating, promotes avid uptake of glucose into muscle, liver, and adipose and stimulates lipogenesis in liver and adipose. Three hours after eating, most of the nutrients from a high-GL meal have been absorbed from the digestive tract (38). However, the persistent anabolic actions from this hormonal response slow the shift from uptake to release of glucose in liver and fatty acids in adipocytes. Consequently, total metabolic fuel concentration in the blood (from glucose, nonesterified fatty acids, and ketones) decreases rapidly in the late postprandial phase, possibly to concentrations below that in the fasting state (39–41). The brain perceives this signal as indicating that critical tissues (e.g., liver) (24) are deprived of energy—a state of “cellular semistarvation” as it has been termed (42)—and may respond to the metabolic challenge with a counter-regulatory hormone response (39). Simultaneously, hunger and cravings for high-GL foods (i.e., those that rapidly raise blood glucose) increase, setting the stage for a vicious cycle. Energy expenditure may also decline related to decreased fuel availability, hormonal (e.g., thyroid) effects on metabolic pathways and thermogenic tissue, or compensatory adaptations (e.g., in autonomic tone) affecting the postprandial state, resting energy expenditure (43), muscular efficiency (44), or physical activity level (45).
Thus, the marked increase in GL across the population since the low-fat diet era—due to a concurrent increase in total carbohydrate and the exceptionally high GI of modern processed carbohydrates (46, 47)—produces a sequence of pathophysiological events that limits metabolic fuel availability to critical oxidative or energy-sensing tissues, especially in the late postprandial phase, driving a positive energy balance (31, 48). Beyond GL, the CIM provides a conceptual framework for understanding how other dietary components [e.g., fructose (49–52), protein and fatty acid type, fiber, food order within a meal (53)], behaviors [e.g., meal timing (54), circadian rhythm, physical activity], and environmental exposures [e.g., endocrine disruptors (55)] may affect body weight through associated mechanisms (e.g., de novo lipogenesis, intestinal function and microbiome, muscle insulin resistance, chronic inflammation, epigenetic modifications) rather than via direct effects on intake and expenditure.
If the CIM is substantially correct, then the strategy to produce a negative energy balance through conscious control of food amount and physical activity level is likely to fail for many people. Restricting energy intake when consuming a high-GL diet would neither lessen the predisposition to fat storage nor diminish hunger during dieting. Rather, by further reducing metabolic fuel availability, hunger would increase and energy expenditure may decline. In contrast, weight reduction produced by carbohydrate restriction would decrease the insulin-to-glucagon ratio, enhance lipolysis and fat oxidation, and result in lower spontaneous food intake. Restriction of carbohydrate intake to very low amounts with consumption of a ketogenic diet may elicit additional mechanisms involving the biological actions of ketones (56), a possibility beyond the scope of this review.
56. Ludwig DS. The ketogenic diet: evidence for optimism but high-quality research needed. J Nutr. 2020;150(6):1354–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Predictions and Testable Hypotheses Arising from the CIM
The mechanistic relations depicted in Figure 1 lead to numerous testable hypotheses (Supplemental Table 2). Key findings from laboratory animal and human research are considered below, according to the corresponding numbers in Supplemental Table 2.
1) Hormonal response to GL
The effects of GL on pancreatic hormone and incretin secretion are well established. As reviewed by Unger (35) 50 y ago, carbohydrate increases the insulin-to-glucagon ratio by not only stimulating insulin secretion, but also paracrine suppression of glucagon secretion. After a 60% compared with 20% carbohydrate meal, the integrated insulin-to-glucagon ratio was 7-fold higher in adults habituated to a high- compared with a low-carbohydrate diet (40). GL also affects incretin hormone secretion. Oral glucose or a high-GI meal rapidly stimulates GIP secretion, produced in the proximate small intestines; conversely, a low-GI meal has a greater effect on glucagon-like peptide-1 (GLP-1), produced in the distal intestines (36, 37).
2) Insulin and tissue-specific insulin sensitivity
Insulin sensitivity is commonly measured at the whole-body level, obscuring critical differences among tissues. Jeanrenaud and colleagues (57, 58) infused rats with insulin by minipump, resulting in well-tolerated hypoglycemia. By day 4 of infusion, glucose utilization, glucose transporter type 4 (GLUT4) expression, lipoprotein lipase activity, and lipogenesis were increased in adipose tissue, whereas muscles developed insulin resistance. These contrasting effects persisted when hypoglycemia was prevented by glucose infusion. Dallon et al. (59) showed that insulin administration reduced uncoupling protein-1 (UCP-1) and mitochondrial respiration in brown adipose tissue. Within the physiological range, insulin regulates energy expenditure (60) at least in part by suppressing UCP-1 mRNA, via peroxisome proliferator–activated receptor γ (PPARG) upregulation, in brown and white adipose tissues in mice—a mechanism that has been recapitulated in cultured 3T3L1 preadipocytes (61).
3) Tissue-specific insulin sensitivity and fat storage
Greater insulin sensitivity in white adipose tissue relative to metabolically active or energy-sensing tissues will alter substrate partitioning in favor of fat deposition. In support of this prediction, mice with adipose-specific ablation of the insulin receptor have reduced adiposity without change in energy intake; they are also protected against age-related metabolic abnormalities (62). Conversely, ablation of the insulin receptor in muscle or the central nervous system increases adiposity (63, 64).
62. Bluher M, Kahn BB, Kahn CR. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science. 2003;299(5606):572–4. [DOI] [PubMed] [Google Scholar][Ref list]
63. Bruning JC, Gautam D, Burks DJ, Gillette J, Schubert M, Orban PC, Klein R, Krone W, Müller-Wieland D, Kahn CR. Role of brain insulin receptor in control of body weight and reproduction. Science. 2000;289(5487):2122–5. [DOI] [PubMed] [Google Scholar][Ref list]
64. Kim JK, Michael MD, Previs SF, Peroni OD, Mauvais-Jarvis F, Neschen S, Kahn BB, Kahn CR, Shulman GI. Redistribution of substrates to adipose tissue promotes obesity in mice with selective insulin resistance in muscle. J Clin Invest. 2000;105(12):1791–7. [DOI] [PMC free article] [PubMed] [Google Scholar][Ref list]
4) Insulin, glucagon, and adiposity
In rodents, chronic insulin treatment increases food intake and adiposity, and these effects are dissociable. Torbay et al. (65) gave rats daily insulin or saline injections, keeping food intake the same between groups. After 4 wk, the insulin-treated rats had increased fat mass and reduced carcass protein. Consistent with this finding, mice with genetically reduced insulin secretion have higher energy expenditure and are protected from diet-induced obesity (61, 66, 67).
66. Templeman NM, Skovso S, Page MM, Lim GE, Johnson JD. A causal role for hyperinsulinemia in obesity. J Endocrinol. 2017;232(3):R173–83. [DOI] [PubMed] [Google Scholar][Ref list]
67. Page MM, Skovso S, Cen H, Chiu AP, Dionne DA, Hutchinson DF, Lim GE, Szabat M, Flibotte S, Sinha Set al. Reducing insulin via conditional partial gene ablation in adults reverses diet-induced weight gain. FASEB J. 2018;32(3):1196–206. [DOI] [PMC free article] [PubMed] [Google Scholar][Ref list]
In humans, insulin and drugs that increase insulin secretion or adipose insulin sensitivity cause weight gain, whereas those that decrease insulin secretion [including α-glucosidase inhibitors (68), which functionally lower the GI of starch] cause weight loss (69). Patients with type 1 diabetes undergoing intensification of insulin therapy for 2 mo experienced a 2.4-kg increase in body fat, associated with decreased energy expenditure and therefore not fully attributable to changes in dietary intake and urinary glucose excretion (70). Genetic variants associated with increased insulin secretion predict weight gain, including in a bidirectional Mendelian randomization study (71, 72). Pancreatic tumors that secrete insulin may cause weight gain, whereas those that secrete glucagon cause weight loss (73).
68. Cai X, Han X, Luo Y, Ji L. Comparisons of the efficacy of alpha glucosidase inhibitors on type 2 diabetes patients between Asian and Caucasian. PLoS One. 2013;8(11):e79421. [DOI] [PMC free article] [PubMed] [Google Scholar][Ref list]
69. Huang Z, Wang W, Huang L, Guo L, Chen C. Suppression of insulin secretion in the treatment of obesity: a systematic review and meta-analysis. Obesity. 2020;28(11):2098–106. [DOI] [PubMed] [Google Scholar][Ref list]
70. Carlson MG, Campbell PJ. Intensive insulin therapy and weight gain in IDDM. Diabetes. 1993;42(12):1700–7. [DOI] [PubMed] [Google Scholar][Ref list]
71. Astley CM, Todd JN, Salem RM, Vedantam S, Ebbeling CB, Huang PL, Ludwig DS, Hirschhorn JN, Florez JC. Genetic evidence that carbohydrate-stimulated insulin secretion leads to obesity. Clin Chem. 2018;64(1):192–200. [DOI] [PMC free article] [PubMed] [Google Scholar][Ref list]
72. Le Stunff C, Fallin D, Schork NJ, Bougnères P. The insulin gene VNTR is associated with fasting insulin levels and development of juvenile obesity. Nat Genet. 2000;26(4):444–6. [DOI] [PubMed] [Google Scholar][Ref list]
73. Wermers RA, Fatourechi V, Wynne AG, Kvols LK, Lloyd RV. The glucagonoma syndrome. Clinical and pathologic features in 21 patients. Medicine (Baltimore). 1996;75(2):53–63. [DOI] [PubMed] [Google Scholar][Ref list]
5) GIP-dominant incretin secretion and fat storage
The cardiometabolic benefits, including weight loss, of GLP-1 agonism are widely recognized. Recently, the adverse effects of GIP*, the incretin with the most potent influence on insulin secretion (74), have come to attention, motivating a search for therapeutic antagonists. GIP administration promotes preadipocyte differentiation and fat storage, and, in the brain, inflammation and insulin resistance. GIP receptor knockout mice are protected against diet-induced obesity (36, 75–78).
*GIP and GLP-1, the two incretin hormones: Similarities and differences [PubMed Abstract] [Full-Text HTML] [Full-Text PDF]. J Diabetes Investig. 2010 Apr 22;1(1-2):8–23. doi: 10.1111/j.2040-1124.2010.00022.x
6) GL, metabolic fuels, food preference, and energy balance
Consumption of a high- compared with a low-GL meal results in reduced circulating metabolic fuel concentration after 3–5 h (39–41, 79), associated with altered substrate partitioning, reduced energy expenditure, and a counter-regulatory response (31, 39, 41, 80, 81). Furthermore, spontaneous transient declines in blood glucose predict hunger, meal initiation or requests, and energy intake (82–84). Insulin administration, which lowers the concentration of all the metabolic fuels, causes hunger and possibly a preference for high-GL foods (21–23). Indeed, endogenous hyperinsulinemia may mediate the cravings and binge-eating behaviors associated with high-GL foods through similar mechanisms (85, 86). Hunger has also been observed with drugs that block sensing or oxidation of metabolic fuels (87–90), with a preference for carbohydrate in some experimental settings (91, 92).
7) GL, energy balance, and obesity
In animals, high- compared with low-GI diets increase adiposity independently of energy intake, in association with lower physical activity level (45, 93–96). Studies comparing high- with low-carbohydrate diets in animals have produced heterogeneous results (97, 98), an issue considered below.
Most but not all acute studies in humans report reduced hunger or energy intake in the late postprandial state after low- compared with high-GI meals (31, 99). Low- compared with high-carbohydrate diets also increase total energy expenditure after 2–3 wk (100). Over the long term, high-fat diets result in more weight loss than high-carbohydrate diets (16, 101–104), contrary to expectation from considerations of energy density. Furthermore, studies suggest a unique diet–phenotype interaction, in that individuals with high insulin secretion or other abnormalities of glucose homeostasis appear especially susceptible to adverse metabolic responses and weight gain when consuming a high-GL diet (96, 105–110). In ad libitum–fed male monkeys, a higher-GL/high-sugar diet compared with a lower-GL/low-sugar diet increased adiposity without increasing total energy intake through age 20 y (111). However, the chronic effect of GL on body composition in humans, with control for energy intake, has not been determined.
8) Causal direction
In experimental models of obesity, metabolic defects involving substrate partitioning and energy expenditure may manifest without (or before) increased food intake, demonstrating the conceptual plausibility of this fundamental feature of the CIM (60, 112–114). This sequence of events is well established in experimental ventromedial hypothalamic obesity (42, 48, 115–119). Immediately after injury to the ventromedial hypothalamus, hyperinsulinemia develops without systemic insulin resistance, and sympathetic nervous system tone is decreased. Importantly, spontaneous physical activity is reduced during development of ventromedial hypothalamic obesity and increased fat deposition occurs even with restriction of food intake to that of controls. (Prevention of hyperinsulinemia after hypothalamic damage diminishes but does not abolish hyperphagia or increased adipose tissue lipogenesis, demonstrating how other influences on adipocyte metabolism, such as autonomic dysfunction, could alter fuel partitioning and cause obesity consistent with the CIM.) A similar sequence of metabolic events, independent of increased food intake, occurs during development of obesity with insulin administration (65, 120) and with a high-GI diet (45, 93–96). Pragmatic and ethical limitations preclude such experiments in humans. In a prospective observational study and a Mendelian randomization analysis, increased adiposity better predicted decreased physical activity level than the converse (121, 122).
Criticisms
With recent attention to the CIM (123), numerous reviews claim to have “falsified” the model, or otherwise challenged its validity (7, 124–135). However, specific criticisms are based on weak or misleading evidence, leading to the following misinterpretations.
Genetic variants associated with adiposity relate primarily to the central nervous system, not the adipocyte as would be predicted by the CIM (134, 135)
The brain expresses genes controlling virtually all aspects of energy metabolism, beyond those that may affect hunger directly, including through hormonal and autonomic control of pancreatic islets, liver, and adipocytes. The identification of more genes related to BMI expressed in the brain than in the adipocyte provides little information about the downstream pathways or organ systems involved. Indeed, pathway analysis identified genetic susceptibility to obesity as involving “insulin secretion/action, energy metabolism, lipid biology and adipogenesis” (1). Of the hundreds of single-nucleotide polymorphisms known to contribute to population risk of obesity, the affected gene, direction of effect, molecular mechanisms, tissues of action, and integrated physiological mechanism are only well-characterized for a handful. In one extensively studied example, obesity resulting from genetic disruption in leptin signaling is not necessarily mediated exclusively through hunger and energy intake; the ob/ob mouse has markedly increased adiposity even when energy-restricted to prevent excessive weight gain (136).
Metabolic fuel concentrations in the blood are typically high, not low, in obesity (134)
As with many physiological systems, causal mechanisms may be evident only during dynamic changes, not after compensation and re-establishment of a new homeostasis. For instance, sodium balance is negative initially, but not chronically, with thiazide diuretic treatment. Reduced circulating fuel concentrations have been documented early in the development of experimental obesity and several hours after a high-GL meal in humans, as considered above. As insulin resistance develops, metabolic fuel concentrations rise. Moreover, circulating fuels are at best a proxy for substrate sensing and oxidation; these may be dissociated under certain conditions, such as insulin resistance or diabetes. The CIM posits a state akin to “internal starvation”—an acceleration of fasting physiology emerging in the late postprandial period. Studies of cellular metabolism and energy sensing are needed to test this hypothesis.
Rodent studies of high-fat diets contradict the CIM (97)
Low-carbohydrate diets have been reported to both cause (97) and prevent (98) adiposity in rodents, heterogeneity that may relate to confounding aspects of diets beyond GL and nondietary factors such as genetic background. Often, high-fat diets used in the laboratory contain not only high amounts of sugar, but also exceptionally high saturated fat content (94), which in rodents causes hyperinsulinemia, liver and muscle insulin resistance, and hypothalamic inflammation (137–143). The ensuing metabolic dysfunction shifts substrate partitioning toward adipose through mechanisms consistent with, not contrary to, the CIM (Figure 1). Substitution of unsaturated fat for saturated fat in these protocols, or ablation of toll-like receptor signaling, reduces metabolic dysfunction and adiposity (137–143).
Energy intake is not reduced by low- compared with high-carbohydrate diets in some feeding studies (135, 144)
Low-carbohydrate diets have high energy density due to their high fat content. Changes in energy density influence food intake acutely (145), but this effect does not persist (17). Myriad factors of dubious relation to chronic energy balance—such as utensil size (146) or plate color (147)—influence acute consumption. The greater long-term weight loss when consuming high-fat diets than when consuming low-fat diets (16, 101–104) highlights the pitfalls of extrapolating chronic macronutrient effects from studies of a few weeks’ duration.
Energy expenditure is not increased by low- compared with high-carbohydrate diets in some feeding studies (128, 135)
Short-term studies of metabolic outcomes may also produce misleading results, related to adaptive processes after changes in macronutrients. With initiation of a very-low-carbohydrate diet, ketones rise, providing an alternative fuel to the brain, thereby preserving lean mass. However, even with fasting, ketones do not reach a steady state until 3 wk (148); on a ketogenic diet, nitrogen balance may remain negative (indicating lean mass breakdown) for 1 mo (149). An adaptive process of several weeks has also been observed with more moderate, nonketogenic macronutrient changes (150–152). In a recent meta-analysis, low- compared with high-carbohydrate diets slightly reduced energy expenditure in trials < 2.5 wk, but low-carbohydrate diets increased energy expenditure in longer trials (100). Contrary to theoretical concerns for experimental error (135), data from a large 5-mo study demonstrating increased energy expenditure when consuming a low-carbohydrate diet during weight-loss maintenance appear robust (106, 153, 154).
Weight loss is not substantially greater for low- than for high-carbohydrate diets in long-term trials (7, 125, 130, 131, 133, 135)
Most diet trials have used low-intensity interventions to promote behavioral change. Most participants in these studies have difficulty sustaining dietary change, limiting inferences about efficacy. In the Pounds Lost (Preventing Obesity Using Novel Dietary Strategies) study (155), participants reported little difference in macronutrient intakes at 2 y and biomeasures of adherence (serum triglycerides, urinary nitrogen excretion) showed little differentiation between dietary groups. In DIETFITS (Diet Intervention Examining The Factors Interacting with Treatment Success), weight loss was greater when consuming a low-carbohydrate than when consuming a low-fat diet at 3 and 6 mo, a difference that did not persist at 12 mo (156, 157). Of note, the low-fat diet included a focus on reducing refined carbohydrates, and both GI and GL decreased in this diet group. The DIRECT trial (Dietary Intervention Randomized Controlled Trial), in which participants received prepared meals to enhance adherence, reported greater weight loss with an ad libitum low-carbohydrate diet than with a calorie-restricted low-fat diet, which persisted at 2 y (158).
Some populations, such as in Asia, consume high-GL diets, yet they have relatively low rates of obesity
The diets of subsistence farming societies were historically based on inexpensive, high-carbohydrate grains and tubers without the metabolic consequences suggested by the CIM. However, such ecological observations are subject to multiple interpretations, among them that people in these rural societies had high levels of occupational physical activity and restricted food availability, both of which might offset dietary influences on weight gain. Among Chinese with recent access to high-GL/high-sugar diets, obesity and metabolic disease have reached epidemic proportions (159). Beyond macronutrient changes, the “nutrition transition” in developing nations beginning in the 1970s (coincident with the low-fat diet era in the United States and Europe) is typified by replacement of traditional carbohydrates with processed starches and sugars that have a higher GL (160). In any event, many dietary and nondietary factors undoubtedly contribute to these trends.
Straw man argument
Some critics assert that the CIM considers the actions of insulin only in the postprandial period and only in adipose tissue, noting that insulin affects adiposity independently of carbohydrate (135). Clearly, insulin is a multifunctional hormone, secreted in response to numerous dietary and nondietary factors. However, the actions of carbohydrate on metabolism persist well beyond the postprandial state, as demonstrated by the “second meal effect,” wherein the GI of supper influences glucose tolerance the next day (161, 162), and by the effect of a low-carbohydrate diet on fasting insulin (163). Indeed, the CIM considers that substrate partitioning and fat deposition are determined by the integrated actions of insulin, together with other hormones and autonomic inputs, in multiple organs, not just adipose tissue (Figure 1). To avoid confusion, it should be recognized that the name of a scientific model typically reflects major distinguishing features—carbohydrate and insulin, here—not the full scope of causal factors and mechanistic relations.
Conversely, one might ask whether our formulation of the EBM is a straw man. In support of our argument, Supplemental Table 1 exemplifies the overwhelming dominance of energy balance in textbooks and professional society statements, and the importance of establishing a negative energy balance, chiefly through decreased intake. None of these articles, nor the recent dismissals of the CIM, provide a mechanistically oriented, testable model addressing dietary drivers of obesity, beyond the recurring focus on widely available, inexpensive, energy-dense, highly palatable, processed foods. The metabolic effects of diet, such as on substrate partitioning—a key distinguishing aspect of the CIM—do not feature in these iterations of the EBM. Illustrating this point, a scientific statement from the Endocrine Society on obesity pathogenesis concludes: “The impact of diet on obesity risk is explained largely by its effect on calorie intake, rather than by changes of either energy expenditure or the internal metabolic environment. Stated differently, ‘a calorie is a calorie.’ Thus, habitual consumption of highly palatable and energy-dense diets predisposes to excess weight gain irrespective of macronutrient content” (7).
Some might argue that new iterations of the CIM serve to “move the goalposts,” obscuring the contest of opposing ideas. However, the relevant question is whether developmental stages of a model offer a consistent distinction from conventional thinking. For the CIM, a clear distinction, implicit in iterations dating back a century (Table 1), entails reversal of the causal pathway linking diet, and specifically carbohydrates, to weight gain.
Resolving controversies
The definitive research needed to resolve persisting controversies will be challenging. The average increase in body weight throughout the last half century is attributable to the storage of <1 g extra fat per day, illustrating the difficulty in exploring causal mechanisms with short-term studies. But this difficulty is no justification for basing scientific knowledge on inconclusive research (135). The failure of the low-fat diet for obesity, as advocated in the late 20th century, can be traced in part to reliance on weak evidence from short-term and confounded studies. Better funding for nutrition research and creative study designs will be needed, including for 1) mechanistically oriented feeding studies of sufficient duration to distinguish transient from chronic macronutrient effects (≥1 mo), focusing on determinants of individual predisposition to dietary effects; 2) clinical trials of efficacy with sufficient treatment intensity and fidelity to promote long-term behavior change (≥1 y); and 3) cohort studies, ideally beginning in childhood, of the natural history of obesity (≥10 y).
Clinical and Public Health Translation
Calorie restriction for obesity treatment results in weight loss—initially—giving patients the impression they have conscious control over their body weight. But predictable biological responses oppose weight loss, including decreased metabolic rate and elevated hunger. Therefore, ongoing weight loss requires progressively more severe calorie restriction, even as hunger increases. Few people achieve clinically significant weight loss over the long term with this approach. Those who cannot might feel implicitly stigmatized as lacking in self-control.
Translation of the EBM to public health may be especially problematic. To prevent obesity, the USDA advises Americans to “stay within calorie limits” (4). Outside the research laboratory, there is no feasible way to measure individual energy requirements to a precision within 300 kcal/d. An overestimation of this magnitude would produce rapid weight gain. For practical purposes, people must determine their dietary energy allowance empirically, as the amount with which body weight remains stable—yet another tautology.
According to the CIM, humans in the modern, industrial food environment may have greater long-term control over what than how much they eat. By reducing anabolic drive with a low-GL diet, patients may experience less hunger and improved energy level, promoting spontaneous weight loss in the same way that an antipyretic reduces fever without conscious control of heat balance. A practical strategy is to substitute high-GL foods (refined grains, potato products, concentrated sugars) with high-fat foods (e.g., nuts, seeds, avocado, olive oil), allowing for moderate intake of total carbohydrate from whole-kernel grains, whole fruits, and legumes and nonstarchy vegetables. For those with special susceptibility, such as high insulin secretion or severe insulin resistance, stricter reduction in total carbohydrate may be optimal.
Conclusions
As with virtually all models of complex biological phenomena, the iteration of the CIM presented here cannot provide a complete and precise representation of all causal mechanisms; nor does it preclude the existence of other causative influences. The value of a scientific model is in stimulating discourse and informing the design of research. Premature claims to have falsified or refuted the CIM, based on weak and confounded evidence, impede constructive scientific discourse. Controversy notwithstanding, important common ground may already exist. For instance, hardwired hedonic preferences for sweetness may drive consumption of sugary foods (consistent with the EBM), which in turn may also affect substrate partitioning through calorie-independent mechanisms (consistent with the CIM). In this sense, conventional notions of palatability and the metabolic effects of preferred foods would work in concert to drive fat accumulation. Or perhaps time-restricted eating could reduce hunger and thereby facilitate calorie restriction, in part through hormonal mechanisms (54).
54. de Cabo R, Mattson MP. Effects of intermittent fasting on health, aging, and disease. N Engl J Med. 2019;381(26):2541–51. [DOI] [PubMed] [Google Scholar][Ref list]
The field of obesity should embrace paradigm clash as an essential step forward. Toward this end, investigators should, first, refrain from hyperbolic claims to have disproven (or proven) alternative explanations of the obesity pandemic; second, clarify the EBM, specifying contrasting causal and testable hypotheses; third, form collaborations among scientists with diverse viewpoints to test predictions in rigorous and unbiased research; and fourth, to facilitate these aims, depersonalize the debate, scrupulously avoiding ad hominem argument. Rigorous research using complementary designs will be needed to resolve the debate, clarify a middle ground, or point the way to new explanatory models that better encompass the evidence. With the massive and growing burden of obesity-related diseases throughout the world, this work must assume priority.