Today, I review, link to, and excerpt from The Cribsiders‘ #100: Cystic Fibrosis – Meet Elexacaftor, Tezacaftor, and Ivacaftor.* December 20, 2023 | By Sam Masur
* St. Onge I, Raymond-Kolker B, Masur S. “#100: Cystic Fibrosis – Meet Elexacaftor, Tezacaftor, and Ivacaftor”. The Cribsiders Pediatric Podcast. https:/www.thecribsiders.com/ December 20, 2023.
All that follows is from the above resource.
AUDIO
Summary:
Check out this milestone episode as we return to Cystic Fibrosis from a more pediatric perspective! Don’t sweat it as Dr. Ina St. Onge returns to walk us through newborn screening, early clinic visits, and the nuances of CFTR modulator therapy.
Cystic Fibrosis Pearls
- The most common symptoms for infants are GI rather than sinopulmonary, specifically poor weight gain.
- Have a low threshold to “sweat” kids (i.e. perform sweat testing) because it’s a cheap, easy, and non-invasive test
- CFTR Modulators can be started as early as 3-6 months of age.
- Pancreatic enzyme replacement therapy (PERT) is started immediately. Even infants are given small spoonfuls of solids in order to take their enzyme replacement (prior to each feed).
- Pulmonary clearance regimen (see below) is recommended to be done 1-2 times per day when feeling well, and doubled when feeling sick.
- Indications for hospitalization include failed outpatient treatment and CF related emergencies, such as large volume hemoptysis due to bronchiectasis, pneumothorax, hyponatremia/dehydration (especially in infants), and bowel obstruction.
- Dr. St. Onge treats CF related bowel obstruction very seriously. If the patient has not had a stool in 2-3 days, the bowel regimen will be aggressively increased like a colonoscopy prep.
Cystic Fibrosis Show Notes
Presentation
Definition
Cystic Fibrosis (CF) is a genetic condition affecting salt transport throughout the body, more specifically through chloride transport. If chloride is unable to move, then salt is unable to move, and thus water is unable to follow that salt, leading to dehydrated secretions throughout the entire body. The most common organs affected include the sinopulmonary tract, GI tract, liver, pancreas, and reproductive tract.
Symptoms
Common symptoms for cystic fibrosis include poor weight gain, recurrent sinopulmonary infections, malabsorptive symptoms like steatorrhea, nasal polyps, or severe constipation (with rectal prolapse). The most common symptoms for infants are GI rather than sinopulmonary, specifically poor weight gain.
Dr. St. Onge recommends having a low threshold to “sweat” kids (i.e. perform sweat testing) because it’s a cheap, easy, and non-invasive test (see below).
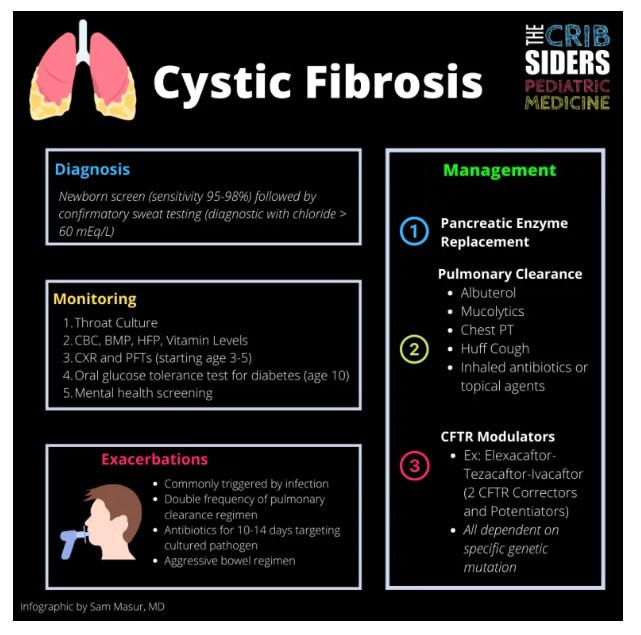
Diagnosis
Newborn Screen
Although state-specific, every newborn screen includes a cystic fibrosis screen. The testing ranges from 95-98% sensitive (Sontag 2009). All babies with positive newborn screens should be referred for sweat testing.
Dr. St. Onge points out that this screen is not 100% sensitive. Therefore, there are plenty of sweat tests ordered on patients with a normal newborn screen.
Sweat Test
Pilocarpine iontophoresis, also known as sweat chloride testing, is performed by placing a device on a patient’s arm. The device has a topical medication on it, which causes sweating, and the sweat is collected in the device. The sweat is sent to the lab, where the chloride content is reported. Prior to administration of the device, there are specific instructions to follow to yield best results, including avoiding lotions, creams, etc.
Values less than 30 mEq/L mean CF is unlikely. Values greater than or equal to 60 mEq/L are considered diagnostic for CF (assuming it has been repeated and still positive). Values between 30 and 59 are considered intermediate or indeterminable. For those with values > 30 mEq/L, Dr. St. Onge recommends repeating the test in a few weeks, but pulmonology will usually coordinate. If still intermediate, these patients may have CF-related metabolic syndrome, which is a disease process with some manifestations of CF. However, all patients with persistently positive or intermediate testing will also have genetic testing sent.
Genetic Testing
Although persistently positive sweat testing is considered diagnostic for cystic fibrosis, genetic testing is still completed because it determines treatment options (see below). Genetic testing includes the most common genetic mutations for cystic fibrosis, including delta F508.
Treatment
Cystic Fibrosis is a multi-disciplinary disease process requiring a team to provide the best care possible. The CF foundation recommends the treatment team includes a pulmonologist, gastroenterologist, endocrinologist, nurse coordinator, dietician, social worker, respiratory therapist, psychologist, and pharmacist.
Baseline Testing Upon Diagnosis
- Genetic testing (usually already sent during sweat testing)
- Fecal elastase to evaluate for pancreatic exocrine insufficiency
- Throat culture (proxy for sputum culture in infants)
- Chest X-Ray within first 6 months
- CBC, BMP, HFP, Vitamin Levels (ADEK)
GI Manifestations
Although infants can show early signs of lung inflammation (Ranganathan 2017), growth is the most important part of CF management as infants. Without overall growth, the lungs won’t grow either. And so the early visits focus on GI manifestations of CF.
Pancreatic enzyme replacement therapy (PERT) is started immediately. Even infants are given small spoonfuls of solids in order to take their enzyme replacement (prior to each feed). They will also receive ADEK vitamins in CF specific formulations. Infants are also started on sodium chloride supplements and a bowel regimen (polyethylene glycol).
Pulmonary Manifestations
Airway clearance is also started immediately, but gradually ramps up. For infants, the CF foundation recommends chest PT +/- albuterol. But hypertonic saline and/or dornase alpha are added as children get older. For older children, the classic airway clearance regimen is as follows (order matters):
- Albuterol. Goal is to bronchodilate before other bronchospastic treatments
- Mucolytics. Either hypertonic saline (HTS) 7% or 3% for mucous hydration. The goal is 7%, but can be switched to 3% if too irritating. After 20 min, patients get dornase alpha to break down the neutrophils in the mucous. The wait is recommended because HTS may inactivate dornase.
- Chest PT done by whichever device is best for each patient
- Huff cough to clear mucous
- Inhaled antibiotics or topical agents. These are used once the airway has been cleared.
This regimen is recommended to be done 1-2 times per day when feeling well. As for the antibiotics, they are strictly to be used based on cultures. However, antibiotics will only be started during exacerbations – the goal of the cultures is to know which bacteria to target. The CF foundation does not recommend preventative antibiotics.
Patients will also be on nasal hydration/rinses because of severe sinus disease as well.
Disease Modifying Agents
CTFR Modulators are the mainstay of treatment for the underlying disease. Underlying genetics will determine where chloride transport fails. The CFTR protein is an ion channel that must be made successfully, then transported to the cell membrane, and then able to function – and of course, any of these steps can go wrong.
CFTR Correctors help protein fold, traffic, and stay on the membrane. CFTR Potentiators help the channel stay open so chloride can move through. The current FDA approved medications are as follows:
- Ivacaftor (CFTR Potentiator)
- Lumacaftor-Ivacaftor (CFTR Corrector-Potentiator, but only available for D5F08)
- Tezacaftor-Ivacaftor (CFTR Corrector-Potentiator)
- Elexacaftor-Tezacaftor-Ivacaftor (2 CFTR Correctors and Potentiators)
These oral medications can be started as early as 3-6 months with Ivacaftor by itself approved down to as young as 2 months of age. These new modulators have extended life expectancy significantly – into the mid 50s and perhaps even further from the mid 30s (Lopez 2023).
Dr. St. Onge says anecdotally there is amazing adherence to these medications because patients feel so much better on them. With the advent of these modulator therapies, researchers are even studying which medications can be removed from CF regimens (SIMPLIFY trial 2023)
Monitoring
Patients are seen every 3 months with their cystic fibrosis care team. The tests that are performed routinely are as follows:
- Throat culture every visit
- Pulmonary function testing (as early as age 3, but most by age 5) every visit
- Annual CBC, BMP, HFP, and vitamin levels
- After age 10, annual oral glucose tolerance test to screen for diabetes. Hemoglobin A1c is not sensitive enough in these patients
- Chest Xray every 1-2 years
- DEXA screen in teenage years
- Mental health screening
- Annual ophthalmologic exam due to risk of cataracts with CFTR modulators
Dr. St. Onge follows liver function closely while on CFTR modulators. Because these medications are so effective, many families choose to stay on the medications despite liver injury.
Exacerbation
At the start of any respiratory illness, Dr. St. Onge recommends increasing the baseline respiratory clearance to a sick plan. So for those performing airway clearance approximately 1-2 times per day, they will increase that number to 3-4 times per day. Dr. St. Onge will try that for a few days, but if there is no improvement, she will add an outpatient oral antibiotic targeted to the known culture data – most commonly targeting MSSA or H. Flu. Although some patients are on inhaled antibiotics already, they are used solely for suppression and are not strong enough to treat an exacerbation – similarly to why we don’t use inhaled antibiotics for community acquired pneumonia.
Indications for hospitalization include CF related emergencies, including large volume hemoptysis due to bronchiectasis, pneumothorax, hyponatremia/dehydration (especially in infants), and bowel obstruction, as well as failed outpatient treatment.
Dr. St. Onge treats CF related bowel obstruction very seriously. If the patient has not had a stool in 2-3 days, the bowel regimen will be aggressively increased like a colonoscopy prep. If the patient begins to vomit, then they should be imaged immediately.
Inpatient Pulmonary Treatment
Once admitted, these patients get more intensive airway clearance regimen and antibiotics as outlined below:
- Airway Clearance with Respiratory Therapy (and vest) 4-5 times per day
- IV antibiotics directed at known pathogens and pseudomonas for 10-14 day course
- IV fluids
There is no consensus on the optimal duration of admission, but it’s roughly 10-14 days based on some adult literature (Goss 2021). Discharge criteria is often based on pulmonary function tests. PFTs will be obtained on admission and then every 3-5 days during the admission. The goal is to return the patient to as close to his/her/their baseline as possible.
Disparities
The landscape of cystic fibrosis is currently changing. African Americans make up approximately 4.1% of CF population, and the hispanic population makes up approximately 9.8%. In these groups especially, there are delays in diagnosis, evaluation, and treatment. Furthermore, the newborn screens have missed some of these groups because they may have different genetic mutations. And with different genetic mutations, some of our most effective medications (CFTR modulators) are unavailable for these patients.
Links