
Amyotrophic Lateral Sclerosis, Resources (1), from emedicine.medscape.com is an excellent brief review 0f ALS for primary care clinicians. What follows are excerpts from those resources.
The following are excerpts from Resource (1):
Amyotrophic lateral sclerosis (ALS) is the most common degenerative disease of the motor neuron system. Although ALS is incurable and fatal, with median survival of 3 years, treatment can extend the length and meaningful quality of life for patients.
Signs and symptoms
In 75-80% of patients, symptoms begin with limb involvement. Initial complaints in patients with lower limb onset are often as follows:
Initial complaints with upper limb onset include the following:
With bulbar onset (20-25%), initial complaints are as follows:
Emotional and special cognitive difficulties in some ALS patients are as follows:
Features of more-advanced disease are as follows:
Progression of bulbar disease leads to the following:
Diagnosis
Definitive diagnosis may not be possible with early ALS. Confirmation of the disease may require a period of observation to document its progressive nature and to exclude alternative diagnoses.
The World Federation of Neurology (WFN) has developed a diagnostic algorithm that combines the clinical and, in some cases, electrophysiologic findings. [1] The degree of certainty of diagnosis is increased by the number of body segments that demonstrate upper motor neuron (UMN) and lower motor neuron (LMN) signs. UMN signs are mild weakness, spasticity, and abnormally brisk reflexes; LMN signs are progressive weakness, wasting, and loss of reflexes and muscle tone.
Management
American Academy of Neurology recommendations for management of patients with ALS can be summarized as follows [2, 3] :
Invasive ventilatory support, requiring tracheostomy, may be considered in the following cases:
Background
Amyotrophic lateral sclerosis (ALS) is the most common degenerative disease of the motor neuron system. The disorder is named for its underlying pathophysiology, with “amyotrophy” referring to the atrophy of muscle fibers, which are denervated as their corresponding anterior horn cells degenerate. “Lateral sclerosis” refers to the changes seen in the lateral columns of the spinal cord as upper motor neuron (UMN) axons in these areas degenerate and are replaced by fibrous astrocytes (gliosis).
The cause of ALS is unknown, although a family history of the disease is obtained in about 5% of patients, and twin studies show a genetic contribution with heritability of about 61%. [10]
Complications
Complications of ALS can include the following:
Diagnosis and Treatment
The diagnosis of ALS is primarily clinical. Electrodiagnostic testing contributes to the diagnostic accuracy (see Clinical Presentation and Workup). Making a diagnosis is important to patients and families, allowing them to stop the search for alternative causes of a patient’s disability and to focus their attention on treatment.
Etiology
Most ALS cases are sporadic, and the specific cause of sporadic ALS is unknown. Many abnormal genes have been identified in familial cases and are considered causal, although the precise mechanism by which they cause ALS is unknown for most.
Occurrence in the United States
Approximately 5600 people in the United States are diagnosed with ALS each year. The annual incidence is 2-3 per 100,000 population; this is about equal to that of multiple sclerosis and 5 times higher than that of Huntington disease. It is estimated that as many as 18,000 Americans may have ALS at any given time.
The lifetime risk for developing ALS for individuals aged 18 years has been estimated to be 1 in 350 for men and 1 in 420 for women. [112] These estimates are close to those reported from 4 European registries, using different methods. [135, 136,137]
Onset of ALS may occur from the teenage years to the late 80s; the incidence rises with increasing age until approximately age 75-80 years. Mean age of onset of sporadic ALS is 65 years; mean age of onset of familial ALS ranges from 46-55 years.
Prognosis [Link is to the the emedicine page that discusses prognosis, progression, and discussion with the patient. What follows below, like this whole post, are simply excerpts.]
ALS is a fatal disease. Median survival is 3 years from clinical onset of weakness. However, longer survival is not rare. About 15% of patients with ALS live 5 years after diagnosis, and about 5% survive for more than 10 years. Long-term survival is associated with a younger age at onset, being male, and limb (rather than bulbar) symptom onset. Rare reports of spontaneous remission exist. [144] In familial ALS that results from an alanine-to-valine mutation in codon 4 of the SOD1 gene (A4V mutation), average survival is 12 months from disease onset. [81]
Frontotemporal executive dysfunction may precede or follow the onset of ALS, but most patients with ALS do not have overt dementia, and cognitive impairment is usually subtle. [146] Approximately 15% of patients with ALS meet criteria for frontotemporal dementia (FTD). Patients with ALS associated with FTD have shorter survival than do those with ALS alone. [147, 148]
Amyotrophic Lateral Sclerosis Differential Diagnoses [This link is to the complete list of diff dxs of ALS from the article. And is included below]:
Diagnostic Considerations
At times, the early presentation of several other neurologic conditions may overlap that of amyotrophic lateral sclerosis (ALS). Appropriate evaluation can exclude these alternatives and confirm the diagnosis of ALS. Fully expressed ALS usually cannot be mistaken for any other disorder.
For patients with a new focal presentation, the differential diagnoses by region include the following:
Upper motor neuron (UMN) bulbar signs: Brainstem lesions including syrinx, mass, stroke, and demyelinating forms of other degenerative diseases
Lower motor neuron (LMN) bulbar signs: Cranial nerve palsies
Limb UMN signs: Cervical myelopathy, cord tumor, hereditary spastic paraparesis, transverse myelopathy, HIV-related myelopathy, syrinx
Limb LMN signs: Radiculopathy, plexopathy, neuropathyDifferential diagnoses for patients with more advanced disease most commonly include the following:
If the onset is rapid (over hours, days or a few weeks), consider disorders such as myasthenia gravis, Guillain-Barré syndrome, acute motor axonal neuropathy, West Nile virus, and botulism.
Other problems to consider, as appropriate, include the following:
Differential Diagnoses
Lyme Disease
CT Scanning and MRI
Brain or spinal MRI may be done to rule out structural lesions and neurologic conditions that sometimes account for early clinical features seen in patients suspected of having ALS (eg, multiple sclerosis, brainstem strokes, tumors, spinal radiculopathy). Results of these studies generally are normal in patients with ALS.
The value of positron emission tomography (PET) scanning and functional MRI in ALS is being investigated. Imaging studies may not be necessary in patients presenting with advanced disease.
Laboratory Studies
Laboratory tests sometimes ordered in the evaluation of a patient with possible ALS include anti-ganglioside M1 (anti-GM1) antibodies, as these can be seen in patients with multifocal motor neuropathy with conduction block. Vitamin B12 and folate levels, HIV status, Lyme serology, and creatine phosphokinase (CPK) determinations may also be performed when indicated by clinical circumstances. The CPK level may be elevated in ALS, but this is not a diagnostic finding.
The following tests may also be considered:
Urinary 24-hour collections for heavy metals may be requested if there is reason to suspect recent exposure. Hexosaminidase A in urine may be checked when adult Tay-Sachs is suspected strongly.
Lyme disease serology may be considered if clinical data suggest that the patient had untreated Lyme disease. However, the history, rather than laboratory testing, drives the diagnosis in Lyme disease.
Genetic testing
In patients with familial ALS, genetic testing may be requested after appropriate counseling. The results of genetic testing may affect not only the patient, but family members as well.
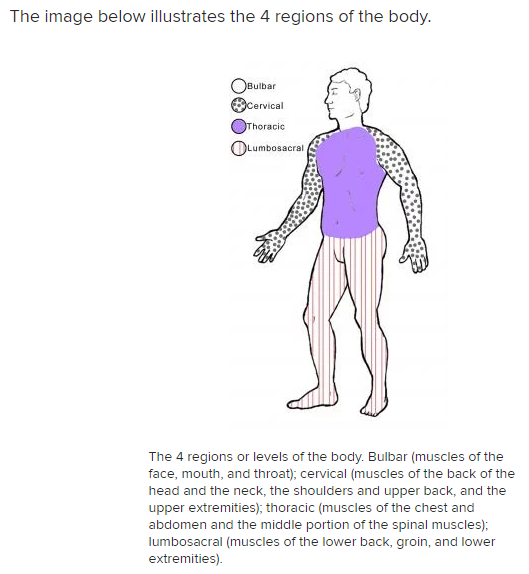
Resources:
Amyotrophic Lateral Sclerosis, Diseases/Conditions, Updated: May 22, 2017
Author: Carmel Armon, MD, MSc, MHS, from emedicine.medscape.com



