
In this post, I link to and excerpt from PedsCases, Cystic Fibrosis: Early Presentation (Part 1 of 2), by Dr. Rose Sun, Oct. 26, 2021.
All that follows is from the above resource.
This podcast is part one of two podcasts that explore cystic fibrosis (CF) in children. This episode focuses on the early presentations of CF and reviews the pathophysiology and etiology of CF in newborns, the presentation and diagnosis of CF in newborns, as well as describes the Newborn CF screening all through a case-based approach. This podcast was developed by Dr. Rose Sun, a second-year Paediatrics resident at the Hospital for Sick Children in Toronto, with the support of Dr. Tamizan Kherani, a pediatric respirologist at the Stollery Children’s Hospital in Edmonton, and Dr. Mariam Ayed, a neonatologist and clinical epidemiologist at Farwaniya Hospital and Kuwait University.
Related Content:
What follows is from the script of the above resource.
Review of Key Learning objectives:[from the end of the script]
So to end this podcast let us review some of the key points:
– Meconium Ileus is an early presentation in 20% of CF patients – Early detection of CF may be based on symptoms, family history of CF or a positive newborn screening
– All of the above requires follow up with the gold standard diagnosis of CF, the sweat chloride test
– CF is an autosomal recessive genetic condition that may present as a severe pancreatic insufficient or a milder pancreatic sufficient phenotype
– With early detection it is important to communicate and educate the parents or care givers on the diagnosis, therapy and management or CFClinical Case 1- Meconium Ileus:
Baby Matt is a 2-day-old infant was born at term (GA39wks) following an unremarkable pregnancy. At birth his weight was 2.84 kg (8th percentile) length 45.3 cm (5th percentile) head circumference 33cm (10th percentile). His vitals were all within the normal range with an axillary temperature of 36.8°C, heart rate was 172 bpm, RR 43 respirations/min blood pressure 68/ 40 mm Hg and O2 saturation 96%. He had an APGAR score of 8 and 8 due to a weak cry and slightly limited flexion of the extremities.
He is generally alert and responsive; however, he appears cachectic. He is pink and the HEENT exam was unremarkable, lungs auscultation did not show any abnormal wheezes and crackles and the heart rhythm is regular, S1 and S2 with no murmurs. His abdomen is distended and multiple doughy loops of dilated bowel present on palpation. He has yet to pass his meconium. In addition he had one episode of bilious vomiting
and is having some difficulties feeding.His mother is 30 years old and this is her 2nd pregnancy with a previous spontaneous abortion at 2 months’ gestational age. Baby Matt was conceived naturally. There was no
maternal history of gestational diabetes, hypertension or infections and antenatal Group B strep screening was negative. Medications in pregnancy included iron, folic acid and
calcium supplements with no history of smoking or alcohol during the pregnancy.Both of Matt’s parents are Caucasian. His mother has a family history of hypothyroidism in her mother and sister. While his father’s side of the family has a history of cystic fibrosis with his grandfather passing away at age 38.
So, given this history what is on your differential for neonatal intestinal obstruction or failure to pass meconium? [Emphasis added.]
Well 99% of healthy full-term infants pass their meconium, first stool, within their first 24 hours of life and the remaining should do so by 48 hours. Possible causes include intrinsic developmental defects in the small and large intestine, abnormalities of peristalsis or abnormal intestinal contents, and insults in utero due to maternal use of narcotics or congenital hypothyroidism. One of the most important conditions that
needs to be ruled out on our differential is CF. This is because up to 20% of newborns with CF present with intestinal obstruction related to meconium ileus. [Emphasis added] The small intestinal in a fetus with CF has mucus glands that produce overly thick secretions in utero; therefore, the meconium formed by these infants is abnormally thick and
adherent and forms mucous plugs that cause obstruction.Now let’s go back to baby Matt. How do we confirm if he does indeed have meconium ileus? We can follow up with some radiographic tests.
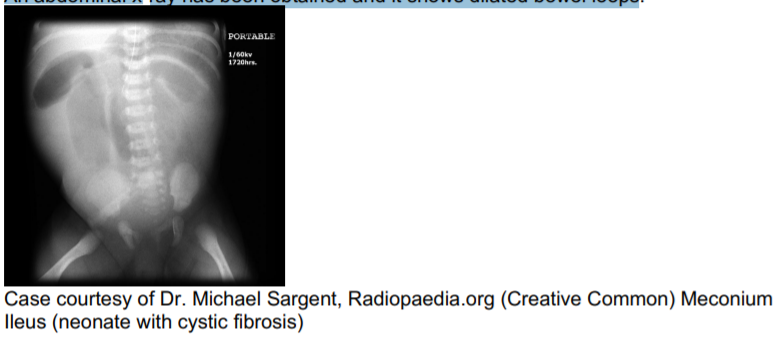
An abdominal x-ray has been obtained and it shows dilated bowel loops.
This was followed by a Lower GI fluoroscopy (Barium Enema) shows a microcolon involving the entire large bowel and impacted meconium pellets in the right colon.
Baby Matt was diagnosed with simple meconium ileus based on both the physical examination and radiological findings.
Meconium ileus can be either simple or complicated. Each occurs with a frequency of approximately 50%.
In the simple form [of meconium ileus], a thick meconium plug begins to form in utero, and because it obstructs the mid-ileum, proximal dilatation, bowel wall thickening, and congestion can occur. Up to 26% of neonates may have abdominal calcifications, although only half are visible on plain radiograph.
In complicated MI, thickened meconium and obstruction lead to complications such as segmental volvulus, atresia, necrosis, perforation, meconium peritonitis, and giant meconium pseudocyst formation.
Now how should we manage baby Matt’s intestinal obstruction?
After birth, both simple and complicated MI should be managed as a newborn intestinal obstruction. Resuscitative measures including respiratory support, if necessary and intravenous
hydration are initiated along with gastric decompression, evaluation, and correction of any coagulation disorders and the inclusion of empiric antibiotic coverage.In the non-operative management of MI, if evacuation is incomplete or if the first attempt at Gastrografin enema evacuation does not reflux contrast into dilated bowel, a second
enema may be necessary. Reflux of the enema into the terminal ileum is critical for the bowel obstruction to be relieved.Serial Gastrografin enemas can be performed at 12- to 24-hour intervals if necessary. Several other wetting agents have been investigated for use as therapeutic enema, but Gastrografin remains the most commonly employed agent. Addition of 1% Nacetylcysteine to the enema solution has been hypothesized to aid in dissolution of the inspissated meconium. The success rate of patients with uncomplicated MI, treated withGastrografin enemas, range from 30% and up to 83%
However, let’s say that the enemas did not work then what should we do next? A more invasive option is to perform a surgical intestinal resection where the rest of the healthy
intestine is sewn back together as a primary anastomosis. Another option is the creation of an ostomy where the two open ends of the intestine are connected to the abdominal wall opening forming a stoma, and the intestines will be washed out and once the obstruction is cleared the intestines will be sewn back together. Baby Matt has undergone the washout enemas and was one of the lucky 30% in which the obstruction
was relieved.Before we get into the confirmation of a CF diagnosis, lets learn a bit more about CF.
Pathophysiology and Prevalence of CF:
CF is an autosomal recessive genetic condition characterized by mutations of the CFTR gene located on chromosome 7. It causes defective chloride channels transport due to cAMP abnormality. The mutations have also been described to cause loss of regulation in epithelial sodium channels. The deregulation in ion transport of epithelial cells, lacking chloride secretion and sodium absorption, leads to insufficient liquid content in
mucus affecting various organs systems including the lungs, gastrointestinal tract, pancreas, liver, and the reproductive tract.The condition is most prevalent in the Caucasian, Northern European populations affecting approximately 1/3300 live births. CF has over 1200 distinct sequences changes that are categorized into 5 classes ranging from severe (class 1-3) pancreatic insufficient to mild (4 &5) pancreatic sufficient. This classification does not correspond to respiratory symptoms.
Delta F508 is the most common mutation accounting for 91% of
CF cases in Canada and is a class 2 mutation It comprises of the deletion of a single phenylalanine residue at amino acid 508. The CFTR gene has low penetrance; therefore, a genotype does not completely dictate the phenotype.Now that we have learned some basics about the etiology, prevalence and pathophysiology of CF. Let’s figure out how to proceed with baby Matt’s care.
Given that there is a positive family history of CF and clinical symptoms of meconium ileus, we can follow up with a sweat chloride test. The sweat test is the gold standard of CF diagnosis through quantitative measurements of electrolytes. A sweat chloride concentration of greater than 60 mmol/L supports a diagnosis of CF whereas a concentration in the intermediate range between 30-60 mmol/L may require retesting and further genotyping. All abnormal sweat tests should be repeated due to the possibly of technical errors such as insufficient sample collection. It is important to also considerpossible false positives influenced by severe eczema, malnutrition, congenital adrenal
hyperplasia and false negatives due to dilution of sample, certain CF mutation variants and peripheral edema. A sweat test may be administered as early as 48 hours of age; however, in newborns with low birth weight such as little Matt there may not be sufficient sweat to sample so we generally wait until they weigh a minimum of 3kg.Little Matt had a sweat chloride test at 7 days of age when he had gained some weight and have sufficient sweat for the test. The first test yielded a chloride concentration of 102 mmol/L and was subsequently repeated with the result of 115 mmol/L. The diagnosis of CF has been confirmed and communicated by his pediatrician to his parents. In later podcasts, we will discuss the long-term management and therapy involved with a diagnosis of CF. I hope you have learned a little about the presentation of meconium ileus, its management and its connection to CF.
Now we will move on to a second case.
Clinical Case 2 – Newborn Screening;
The newborn in this case was diagnosed by newborn screening exam. There is no history of genetic disorders in either parent’s family.
Her parents have consented to newborn screening and dried blood spot specimens from a heel poke were obtained 2 days after the birth. Two weeks later, her results came back and the immunoreactive trypsinogen level (IRT) positive was elevated (99.9th percentile).
The IRT is then followed by a genetic analysis on the same spot of blood to confirm the results. For baby Linda it showed G551D and G542X mutations. In cases like baby Linda, newborns with an elevated IRT and two mutations are reported as having probable CF. A genetic counselor will then contact the ordering or family physician to discuss the positive result usually within 3-4 weeks of the initial screening. The ordering or family physician will then contact the family and refer the child to a specialized pediatric CF clinic. All newborns with a probable or inconclusive CF screen are then referred for sweat chloride testing. At a later date genetic counseling will be offered to extended family members.



