
In this post I link to and excerpt from Genetics of mitral valve prolapse and its clinical impact [PubMed Abstract] [Full Text HTML] [Full Text PDF]. E-Journal of Cardiology Practice – Volume 16, Jan 16, 2019.
Here are excerpts:
Mitral valve prolapse (MVP) is the most frequent valvular abnormality, associated with various degrees of incompetence and sequelae including heart failure and sudden death. MVP can be familial in 35-50% of cases. In addition to concomitant connective tissue syndromes, MVP has an important heritable component demonstrated in community-based studies and confirmed from recent genome-wide association studies identifying several genetic risk loci. Genomic and precision medicine will result in increasing availability of genomic data and interpreting these clinically will be a challenge for clinicians. In this contemporaneous review we summarise MVP genetics and clinical implications.
Introduction
Mitral valve prolapse (MVP) is the most frequent valvular abnormality (prevalence 2-3%) typified by left atrial displacement of either or both of the anterior and posterior leaflets [1]. Although it is usually benign without any serious long-term consequences, in some patients it can result in severe mitral regurgitation, and is associated with heart failure syndrome, arrhythmia and sudden cardiac death [2, 3]. In the majority of cases, MVP is isolated and not associated with other conditions. However, MVP can also be seen as part of other clinical syndromes such as connective tissue disorders and idiopathic dilated cardiomyopathy. A familial pattern is seen in approximately 35-50% of cases, suggesting a heritable component which has prompted genetic research into this condition.
Given that MV prolapse is prevalent and rare events, such as sudden death, have been studied at post-mortem, previously undiagnosed MV prolapse has been noted at a higher frequency in these decedents, suggesting an association [4]. However, this association of MVP with sudden cardiac death (SCD) may in part be due to ascertainment bias. More recent data have identified a plausible link with arrhythmogenicity and a familial predisposition [2, 3]. There are complex variables involved including structural, pathological, electrophysiological and possibly genetic aspects to consider. In this review, we summarise the current understanding of the genetics of MVP and the clinical relevance and impact of these findings.
MVP aetiology and pathophysiology
MVP is defined on imaging by billowing of any portion of the mitral leaflets (≥2 mm above the annular plane on a long-axis view [parasternal or apical 3-chamber]) into the left atrium during systole [5]. The mechanism of prolapse varies and can include disruption or elongation of the leaflets directly or the subvalvular apparatus (chordae or papillary muscles).
Two main forms of MVP exist based on aetiology:
- primary (degenerative disease in the absence of identifiable connective tissue disease and fibroelastic disease); and
- secondary MVP (associated with an identifiable disorder such as connective tissue disease, e.g., Marfan syndrome and Ehlers-Danlos syndrome).
Epidemiology
The epidemiology was initially highly variable in the literature, due in part to poor study design and less stringent definitions of MVP, but is currently estimated at 2.4% (17 million in the USA and 178 million worldwide) [1]. The majority of MVP is benign, but it is associated with varying degrees of regurgitation and with endocarditis, heart failure syndrome and SCD. MVP remains the leading indication for surgery of the MV [7]. MVP exists in sporadic and familial forms, recognised since the 1960s, and has a prevalence of 5-20% in first-degree relatives of probands [8-10 in the excerpt-1, 2, and 3 below].
- Delling FN, Li X, Li S, Yang Q, Xanthakis V, Martinsson A, Andell P, Lehman BT, Osypiuk EW, Stantchev P, Zöller B, Benjamin EJ, Sundquist K, Vasan RS, Smith JG. Heritability of Mitral Regurgitation: Observations From the Framingham Heart Study and Swedish Population. Circ Cardiovasc Genet. 2017 Oct;10(5).
- Kelly RE, Goretsky MJ, Obermeyer R, Kuhn MA, Redlinger R, Haney TS, Moskowitz A, Nuss D. Twenty-one years of experience with minimally invasive repair of pectus excavatum by the Nuss procedure in 1215 patients. Ann Surg. 2010;252:1072-81.
- Strahan NV, Murphy EA, Fortuin NJ, Come PC, Humphries JO. Inheritance of the mitral valve prolapse syndrome. Discussion of a three-dimensional penetrance model. Am J Med. 1983;74:967-72.
Electrocardiographic abnormalities
Abnormalities of the T-waves have been reported in cases of MVP and sudden death or ventricular arrhythmias [2 in the article and 1, below in this excerpt]. These repolarisation abnormalities can reflect endocardial and mid-myocardial changes of papillary muscles or the left ventricle and may result in a repolarisation gradient across the myocardium, resulting in T-wave inversion.
- Basso C, Perazzolo Marra M, Rizzo S, De Lazzari M, Giorgi B, Cipriani A, Frigo AC, Rigato I, Migliore F, Pilichou K, Bertaglia E, Cacciavillani L, Bauce B, Corrado D, Thiene G, Iliceto S. Arrhythmic Mitral Valve Prolapse and Sudden Cardiac Death. Circulation. 2015;132:556-66.
Sudden death risk and MVP
Sudden death was observed in 6 (2.5%) patients in an echocardiographic cohort of 237 patients followed for a mean of 6.2 years (1,469 patient-years), where the only variable associated with SCD was redundant leaflets [8].
Cases of sudden death (some with documented ventricular arrhythmias) were noted to have prolonged QT intervals, particularly among female decedents [4, 11]. In a retrospective review of 24 cases with out-of-hospital cardiac arrests, 10 (42%) had bileaflet MVP and 90% of victims were female [3]. These cases had biphasic and inverted T-waves in the inferior leads on 12-lead ECG and a high frequency of premature ventricular contractions (PVC) on Holter monitoring with an origin from the right ventricular outflow tract, papillary muscle, or were of fascicular origin. In a study by Basso et al, imaging and post-mortem findings in patients with MVP and sudden death were compared to patients with MVP and documented ventricular arrhythmias and controls [2]. The study identified scarring which may have been as a result of increased tension in the chordae which transmit to the subvalvular apparatus, resulting in fibrosis acting as an abnormal substrate. Others have proposed possible mechanical triggers as a result of redundant valve tissue prolapsing to the atrium in systole and ventricle in diastole, increasing the propensity for premature ventricular contractions (PVCs), which may initiate ventricular arrhythmias.
Thus, the presence of T-wave inversion, PVCs, fibrosis on cardiac MRI and a family history of sudden unexplained deaths should raise suspicion of a malignant phenotype.
Genetics
Although familial forms have been recognised for some time, determining the genetic aetiology has been challenging until more recent technological advances. Cascade clinical screening of first-degree relatives is highly variable, especially if there are non-syndromic vs. syndromic associations. Familial forms with classic Mendelian inheritance were detected with moderate success using linkage disequilibrium analyses, followed by candidate gene approaches. More recently, novel variant discovery, using whole exome and whole genome sequencing approaches, has been used to identify single genes as well as single-nucleotide polymorphisms (SNP) through genome-wide association studies (GWAS). The current known genes and SNP are described below and classified into syndromic and non-syndromic forms (Figure 1).
Figure 1. Schematic of the complexities of MVP genetics.
The implicated genes have been organised into those associated with syndromic conditions and a higher frequency of MVP than that seen in the general population, those associated with familial non-syndromic forms, and genome-wide association studies.
The red arrow on the apical 4-chamber 2-dimensional echo tracing in late systole shows the posterior mitral valve leaflet prolapsing into the left atrium.
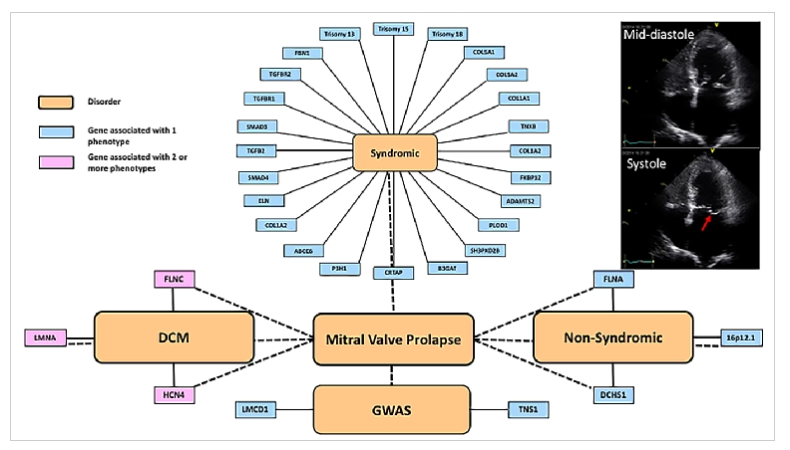
Please see the article for details on the following:
Syndromic and without connective tissue disease
- Trisomies (chromosomes 18, 13, and 15)
Syndromic associated with connective tissue disease
- Marfan syndrome (MFS)
- Loeys-Dietz syndrome (LDS)
- Ehlers-Danlos syndrome (EDS)
- Williams-Beuren syndrome (WBS)
- Pseudoxanthoma elasticum
- Osteogenesis imperfecta
- Larsenlike syndrome
- Borrone dermatio-cardio-skeletal syndrome
- Cardiomyopathies and ion channelopathies (FLNC, LMNA, HCN4)
- Genome-wide association studies
- Non-syndromic myxomatous MVP
Translational outlook
Although syndromic MVP has been well recognised for decades and the role of genetics is more established, the evaluation is usually based on the syndrome and not the MVP itself. Most syndromic forms are autosomal dominant and a minimum 3-generational pedigree, with evaluation at follow-up (to identify new individuals who may not have expressed the phenotype) is standard in most centres. Although familial patterns and heritability have long been recognised, the more recent genetic elucidation of non-syndromic MVP is challenging understanding of the disease. We advocate a 3-generational family history in cases of MVP and clinical cascade screening of first-degree relatives. This is likely to be cost-effective and easily translatable as MVP (defined above) can be identified. The challenge with mild prolapse not meeting current imaging criteria is more demanding; we suggest avoiding this at the present time. We do not advocate cascade genetic screening for non-syndromic MVP at this time, due to limited understanding and data on the validity, reliability and cost-effectiveness.
The widespread availability of clinical and research genetic testing, including commercial kits available direct to the public without a request from a clinician, is likely to result in variants being identified more readily. Interpreting these genetic findings is a challenge for clinical staff and thus enrolment into clinical research studies with access to experts is recommended. We also advocate that genetic testing without full genetic counselling, including implications of test results, should be avoided.
Conclusions
Understanding the genetic aetiology of non-syndromic MVP vs. syndromic MVP has only just begun. The aims are to improve mechanistic understanding, particularly with regard to the pathogenesis, which may explain the varied phenotypic expression and ultimately pave the way for novel therapeutics. Although the genetics of MVP are still largely in the research domain, the key to better understanding is good phenotyping (with robust echocardiographic assessment) and clinical evaluation for associated features. Furthermore, full and accurate pedigree analysis is key to improving the accuracy of epidemiological data, as well as to understanding heritability. There is a recognised association with sudden death and the presence of T-wave inversion. PVCs and fibrosis on late gadolinium enhancement should alert the clinician that the patient may be at risk of malignant arrhythmias and sudden death.



