
This post contains links to and excerpts from (1) How to Interpret and Pursue an Abnormal Complete Blood Cell Count in Adults [PubMed Abstract] [Full Text HTML] [Full Text PDF]. Mayo Clin Proc. 2005 with additional resources after the post.
Here are excerpts from the above article:
Abstract
A complete blood cell count (CBC) is one of the most common
laboratory tests in medicine. For example, at our institution alone, approximately 1800 CBCs are ordered every day, and 10% to 20% of results are reported as abnormal. Therefore, it is in every clinician’s interest to have some understanding of the specific test basics as well as a structured action plan when confronted with abnormal CBC results. In this article, we provide practical diagnostic algorithms that address frequently encountered conditions associated with CBC abnormalities including anemia, thrombocytopenia, leukopenia, polycythemia, thrombocytosis, and leukocytosis. The objective is to help the nonhematologist recognize when a subspecialty consultation is reasonable and when it may be circumvented, thus allowing a cost-effective and intellectually rewarding practice.Abreviations for the article
ACD = anemia of chronic disease; ANC = absolute neutrophil count;
CBC = complete blood cell count; CML = chronic myeloid leukemia;
ET = essential thrombocythemia; FISH = fluorescence in situ hybridization; Hct = hematocrit; HES = hypereosinophilic syndrome; Hgb = hemoglobin; HIV = human immunodeficiency virus; IDA = iron deficiency anemia; ITP = idiopathic thrombocytopenic purpura; LDH = lactate dehydrogenase; LGL = large granular lymphocyte; MCV = mean corpuscular volume; MDS = myelodysplastic syndrome; PA = pernicious anemia; PBS = peripheral blood smear; PT = primary thrombocytosis; PV = polycythemia vera; RBC = red blood cell; RCM = RBC mass; RT = reactive thrombocytosis; TCR = T-cell receptor; TTP/HUS = thrombotic thrombocytopenic purpura/hemolytic uremic syndrome; WBC = white blood cellFor practical purposes, the variables to focus on when
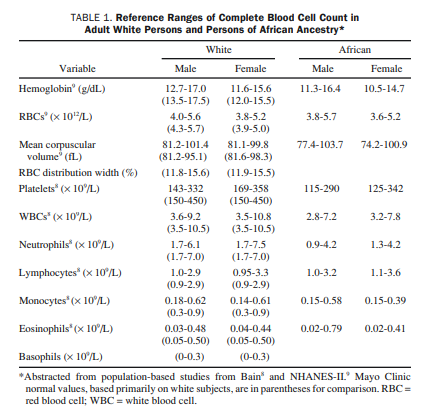
examining the CBC are:
- Hgb (as a general indicator of anemia or polycythemia),
- MCV (a key parameter for the classification of anemias),
- RBC distribution width (a relatively useful parameter in the differential diagnosis of anemia),
- RBC count (an increased RBC count associated with anemia is characteristic in the thalassemia trait),
- platelet count (to detect either thrombocytopenia or thrombocythemia), and
- WBC count with differential (usually gives important clues for the diagnosis of acute leukemia and chronic lymphoid or myeloid disorders as well as for the presence of leukopenia and neutropenia).
Furthermore, in patients with an abnormal WBC count, the clinician should immediately ask which WBC type is affected: neutrophils, lymphocytes, monocytes, eosinophils, or basophils.
In this regard, the machine-derived 5-part differential should be confirmed by the human eye (ie, peripheral blood smear [PBS] examination) before it is acted on.
Anemia
The first step in approaching anemia is to classify the
process as microcytic (MCV, <80 fL), normocytic (MCV,
80-100 fL), or macrocytic (MCV, >100 fL).10,11 This exercise markedly narrows the differential diagnosis that needs
to be considered in each patient. Also, we strongly recommend obtaining a PBS during the initial evaluation of anemia, regardless of subtype. A PBS substantially enhances the initial process of differential diagnosis and provides guidance for further testing.MICROCYTIC ANEMIA
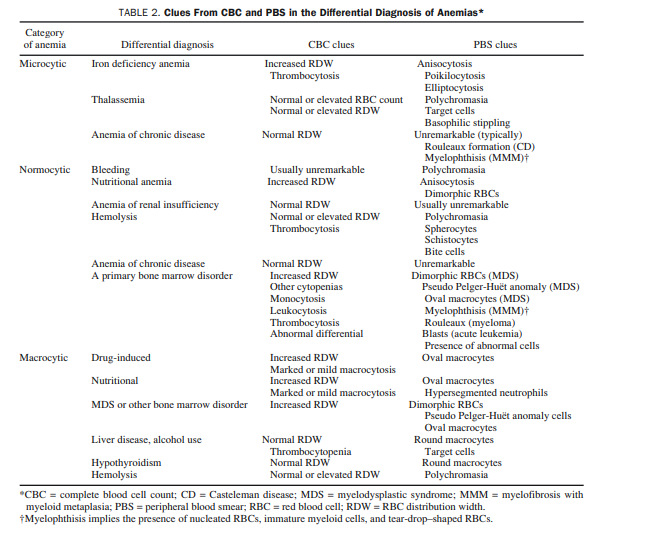
The 3 major diagnostic possibilities for microcytic anemia
are iron deficiency anemia (IDA), thalassemia, and anemia
of chronic disease (ACD).10,11 A fourth possibility,
sideroblastic anemia presenting with microcytosis, is not
prevalent enough for routine consideration.12 Clues from
the CBC and PBS for the differential diagnosis of microcytic anemia are outlined in Table 2.
Since the most common of the microcytic anemias is IDA, we recommend determination of the serum ferritin level as the initial step for all patients with microcytic anemia (Figure 1).13 A low serum ferritin level is diagnostic of IDA. Similarly, contrary to current dogma regarding acute phase reaction, a
diagnosis of IDA is unlikely in the presence of a persistently normal or elevated serum ferritin level.14 In general,
we do not recommend either other serum iron studies (serum iron, total iron-binding capacity, transferrin saturation)
or bone marrow biopsy for evaluation of IDA.10,14
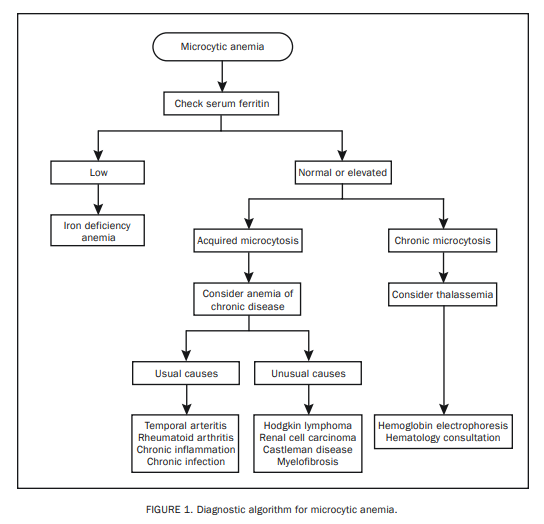
Acquired microcytic anemia that is not IDA is indicative of an underlying systemic disease and is labeled operationally as microcytic ACD.16,17 Both usual and unusual systemic disease may accompany microcytic ACD (Figure 1).18 Further laboratory investigation in this instance as well as the need for a hematology consultation is dictated by patient history and findings from both the physical examination and the PBS.
NORMOCYTIC ANEMIA
The first step in approaching normocytic anemia is to exclude potentially treatable causes from the standpoint of anemia, including bleeding, nutritional anemia, anemia of renal insufficiency,19 and hemolysis (Figure 2). Clues from the CBC and PBS for each of these categories are listed in Table 2.
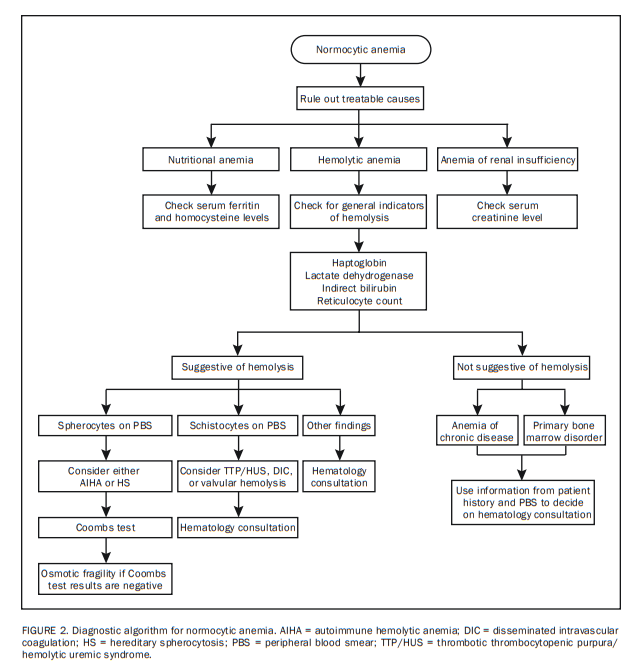
Patient history is key in implicating bleeding as a cause of anemia, and a fecal occult blood test can be ordered if indicated.
Regarding nutritional anemia, it should be noted that both iron and vitamin B12/folate deficiencies are possible causes of “normocytic” anemia, despite their usual association with microcytic and macrocytic anemia, respectively.13,20
Anemia of renal insufficiency is addressed easily by checking the serum creatinine level.
Initial laboratory tests that should be ordered when hemolysis is suspected and/or to exclude the possibility of active hemolysis include serum levels of haptoglobin, lactate dehydrogenase (LDH), and indirect bilirubin as well as reticulocyte count and the PBS (Figure 2).21,22 In general, active hemolysis is suspected if a low haptoglobin level is associated with increased LDH, indirect bilirubin, or reticulocyte count.
The differential diagnosis of a normocytic anemia that is not linked to bleeding, nutrition, renal insufficiency, or hemolysis is either normocytic ACD or a primary bone marrow disorder.17 Patient history and PBS results provide the most helpful information in distinguishing the two (Table 2; Figure 2 [Both included above]).
Here is the part of Table 2 [above] that relates to normocytic anemia:
![]()


And here are links to additional resources on the above from emedicine.medscape.com:
- Castleman disease (CD)
- Myelodysplastic syndrome (MDS)
- Myelofibrosis with myeloid metaplasia (MMM)
- Myelophthisis implies the presenece of nucleated RBCs, immature myeloid cells, and tear-drop-shaped RBCS
In general, in patients with normocytic anemia, a hematology consultation may be unnecessary if the patient history, the initial laboratory test results described previously, and the PBS results are consistent with nutritional anemia, anemia of renal insufficiency, or normocytic ACD.
Furthermore, some PBS results may dictate the ordering of
additional laboratory tests without waiting for approval
from a hematologist: (1) a Coombs test and if results are
negative, an osmotic fragility test for patients with spherocytosis and (2) coagulation, haptoglobin, and LDH tests for patients with schistocytosis (Figure 2).Similarly, a urinary hemosiderin test is extremely helpful if valvular hemolysis is suspected.
All other scenarios require a hematology consultation.
Finally, the possibility of drug-induced hemolysis always must be considered.21
MACROCYTIC ANEMIA
Use of certain drugs (eg, hydroxyurea, zidovudine) and
alcohol consumption are notoriously associated with macrocytosis and should be first considerations during evaluation of macrocytic anemia (Figure 3).23,24*Myelodisplastic Syndrome [Link is to the emedicine.medscape.com article]
The next step is to rule out nutritional causes (B12 or folate deficiency); we prefer to use serum homocysteine for initial screening because of its higher test sensitivity.20,25
However, we advocate concomitant determination of the serum B12 level [along with the serum homocysteine] to safeguard against laboratory error in view of the dire clinical consequences associated with vitamin B12 deficiency
(Figure 3).If 1 of the 2 tests [above] has abnormal results, the serum methylmalonic acid level should be checked; an increased level strongly suggests B12 deficiency.
In patients with vitamin B12 deficiency, the next step is
to screen for the presence of intrinsic factor antibodies26,27;
if present, a working diagnosis of pernicious anemia (PA)
is made. Otherwise, the Schilling test is performed to differentiate PA from primary intestinal malabsorptive disorders.27,28Further investigation of macrocytic anemia that is
neither drug-induced nor nutritional is simplified by
subcategorizing the process into either a marked (MCV, >110 fL) or mild (MCV, 100-110 fL) subtype. In this instance, markedly macrocytic anemia is almost always associated with primary bone marrow disease, whereas mildly macrocytic anemia also can be associated with more benign conditions (Figure 3).29,30THROMBOCYTOPENIA
The first step in treating thrombocytopenia is to exclude the
possibility of spurious thrombocytopenia caused by EDTA-induced platelet clumping (Figure 4).31 The situation is clarified by either examining the PBS or repeating the CBC using sodium citrate as an anticoagulant.Another important point to consider before starting a costly search for disease is the fact that healthy women may experience mild to moderate thrombocytopenia (platelets, 75-150 × 109/L) during pregnancy, and such incidental thrombocytopenia of pregnancy requires no further investigation.32

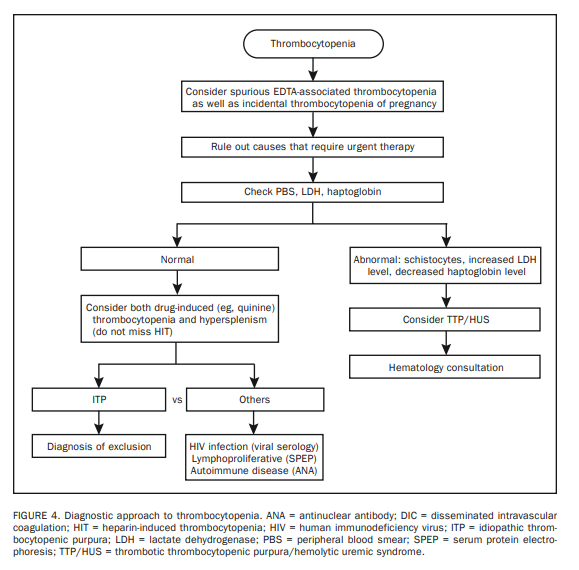
The second step in treating patients with thrombocytopenia is to always consider the possibility of thrombotic thrombocytopenic purpura/hemolytic uremic syndrome (TTP/HUS) because of the urgency for specific therapy for these diagnoses (ie, plasma apheresis).33,34 This is why we recommend PBS (to look for schistocytes); serum levels of haptoglobin and LDH (to assess for concomitant hemolysis) and creatinine; and coagulation tests including plasma levels of D-dimer, in most instances of thrombocytopenia.
Both TTP/HUS and disseminated intravascular coagulation are characterized by microangiopathic hemolytic anemia and thus display schistocytes on PBS, an increased
LDH level, and a decreased haptoglobin level.35 However,
coagulation studies are usually normal in TTP/HUS, whereas clotting times are prolonged in disseminated intravascular coagulation. Regardless, suspected TTP/HUS requires a hematology consultation.The third step is consideration of both drug-related
thrombocytopenia and hypersplenism in all instances.36-38Thrombocytopenia is more likely to occur in the presence
of hypersplenism* associated with cirrhosis.39*Hypersplenism is a common disorder characterized by an enlarged spleen which causes rapid and premature destruction of blood cells. Resource (3) below
*See Hypersplenism: History and current status [PubMed Abstract] [Full Text HTML] [Full Text PDF]. Exp Ther Med. 2016 Oct; 12(4): 2377–2382.
*See also Point-of-care ultrasonography improves the diagnosis of splenomegaly in hospitalized patients [PubMed Abstract] [Full Text HTML] [Full Text PDF]. Crit Ultrasound J. 2015; 7: 13
The most frequently implicated drugs in thrombocytopenia are antiboics including trimethoprim-sulfamethoxazole, cardiac
medications (eg, quinidine, procainamide), thiazide diuretics, antirheumatics including gold salts, and heparin.36Heparin-induced thrombocytopenia is potentially catastrophic
and requires immediate cessation of drug use, including heparin flushes.40 A diagnosis of heparin-induced thrombocytopenia may be confirmed by in vitro testing to detect heparin dependent platelet antibodies.After microangiopathic hemolytic anemia, drug-induced thrombocytopenia, and hypersplenism have been ruled out, idiopathic thrombocytopenic purpura (ITP) becomes the major contender in the differential diagnosis of isolated thrombocytopenia.41-43 However, ITP is currently a diagnosis of exclusion that requires consideration of other causes of immune-mediated thrombocytopenia including connective tissue disease, lymphoproliferative disorders, and human immunodeficiency virus (HIV) infection.44
Therefore, we recommend laboratory tests for HIV*, antinuclear antibodies**, and monoclonal protein*** for further investigation.
*See the following for further information:
- HIV Testing Overview Updated: Sep 19, 2018
Author: David J Cennimo, MD, FAAP, FACP, AAHIVS from emedicine.medscape.com- Rapid HIV Testing Updated: Feb 04, 2019
Author: Jacob D Isserman, MD from emedicine.medscape.com**See the following for further information:
- Antinuclear Antibody Updated: Sep 04, 2014
Author: Asma Al-Zougbi, MD from emedicine.medscape.com- Antinuclear Antibodies: Marker of Diagnosis and Evolution in Autoimmune Diseases [PubMed Abstract] [Full Text HTML] [Full Text PDF]. Lab Med. 2018 Jul 5;49(3):e62-e73. doi: 10.1093/labmed/lmy024.
- Autoimmune Antibodies Updated: Nov 24, 2014 Author: Buck Christensen from emedicine.medscape.com
**See the following for further information:
- Serum Protein Electrophoresis Updated: Jul 31, 2019
Author: Sherilyn Alvaran Tuazon, MD emedicine.medscape.com- Immunofixation Updated: Jul 29, 2019 Author: Anastasios Dimou, MD from emedicine.medscape.com
In contrast, neither platelet antibody test nor bone marrow biopsy is indicated in the work-up of most patients with isolated thrombocytopenia that is consistent with ITP.45
Rare causes of isolated thrombocytopenia include hereditary thrombocytopenias that may be associated with giant platelets on PBS (eg, May-Hegglin anomaly, gray platelet syndrome, Bernard-Soulier syndrome, and Xlinked Wiskott-Aldrich syndrome),46 myelodysplastic syndrome* (MDS) (rarely presents with isolated thrombocytopenia),47 amegakaryocytic thrombocytopenia (a bone marrow biopsy is required for diagnosis),48 and posttransfusion purpura (a rare complication of blood transfusion).49 A history of blood component transfusion at 1 to 2 weeks before onset of thrombocytopenia should suggest posttransfusion purpura.
*See the following for further information
- Myelodysplastic Syndrome Updated: Jul 24, 2018. Author: Emmanuel C Besa from emedicine.medscape.com
In all the aforementioned [above] situations, a hematology consultation is advised.
LEUKOPENIA
NEUTROPENIA
The absolute neutrophil count (ANC) is either derived by
multiplying the total leukocyte count by the percentage of
band neutrophils and segmented neutrophils or obtained
directly from an electronic cell counter. Neutropenia is
clinically most relevant when it is severe (ANC, <0.5 × 109/L) because of the associated risk of infection.50Severe neutropenia is classified into congenital and acquired categories.
Both hematology and medical genetics consultations are advised for patients with congenital severe neutropenia but not for those with benign chronic neutropenia that occurs usually in persons of African or Yemenite Jewish ancestry without sparing other
ethnic groups.52-56 The ANC in benign chronic neutropenia
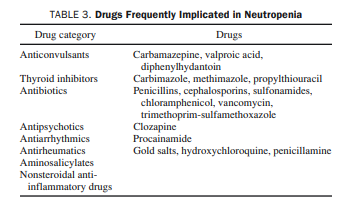
ranges usually between 0.5 × 109 /L and 1.5 × 109/L, and the clinical course is asymptomatic.The most frequent cause of acquired neutropenia is drug
therapy; the most commonly implicated agents are listed in
Table 3.51,57,58 However, any drug should be assumed to be a
potential offender until proved otherwise.
Infection is another common cause of neutropenia, and the major culprits are viruses and sepsis.
In the clinical setting, where either drug- or infection-associated neutropenia is suspected, appropriate immediate measures include discontinuation of the presumed offending agent, close monitoring of daily CBC, and consideration of treatment with a myeloid growth factor* in patients with uncontrolled bacterial or fungal infection.
*For further information see:
- Issues on the Use of White Blood Cell Growth Factors in Oncology Practice [PubMed Abstract] [Full Text HTML] [Full Text PDF]
- Recommendations for the Use of WBC Growth Factors: American Society of Clinical Oncology Clinical Practice Guideline Update [PubMed Abstract] [Full Text HTML] [Full Text PDF]
Other causes of acquired neutropenia include immune neutropenia, large granular lymphocyte (LGL) leukemia,
and other hematologic malignancies that present only rarely
with isolated neutropenia (eg, MDS).51,59-61[The evaluation of the above is discussed in the article but is complex and in many cases would best be pursued with a hematology consult.]
LYMPHOPENIA
The possibility of recent therapy with immunosuppressive
drugs, including corticosteroids and antilymphocyte monoclonal antibodies, must be considered first in treating the
patient with lymphopenia.62,63Other causes of acquired lymphopenia, which should be familiar to the primary care physician, include viral infections such as acquired immunodeficiency syndrome and severe acute respiratory syndrome,64,65 critical illness including sepsis,66 autoimmune and connective tissue diseases including lupus and rheumatoid arthritis,67-69 sarcoidosis,70 chronic renal failure,71,72 excess alcohol use,73 older age,74 thymoma,75 and tuberculosis and other bacterial infections.76
An immunology consultation is advised if congenital lymphopenia is suspected including Bruton X-linked agammaglobulinemia (B-cell deficiency), severe combined immunodeficiency (B-cell and T-cell deficiency), and DiGeorge syndrome (T-cell deficiency).77,78
Regarding common variable immunodeficiency, the most common primary immunodeficiency syndrome that is symptomatic, it is important to know that the lymphocyte count may or may not be normal.79
POLYCYTHEMIA
Resources:
(1) How to Interpret and Pursue an Abnormal Complete Blood Cell Count in Adults [PubMed Abstract] [Full Text HTML] [Full Text PDF]. Mayo Clin Proc. 2005. Jul;80(7):923-36.
(2) #167 LIVE! Common CBC Abnormalities with Mary Kwok MD [Link is to the podcast and show notes]. AUGUST 19, 2019 By MATTHEW WATTO, MD from The Curbsiders.
(3) Hypersplenism: History and current status [PubMed Abstract] [Full Text HTML] [Full Text PDF]. Exp Ther Med. 2016 Oct; 12(4): 2377–2382.
(4) Point-of-care ultrasonography improves the diagnosis of splenomegaly in hospitalized patients [PubMed Abstract] [Full Text HTML] [Full Text PDF]. Crit Ultrasound J. 2015; 7: 13
(5) Approach To Lymphocytosis 2017 from Cancer Therapy Adviser
(6) Evaluation of Patients with Leukocytosis [PubMed Abstract] [Full Text HTML] [Full Text PDF]. Am Fam Physician. 2015 Dec 1;92(11):1004-11.



