
In this post I link to and excerpt from Mitral valve prolapse, a familial condition worth screening for? [PubMed Abstract] [Full Text HTML] [Full Text PDF]. Eur J Prev Cardiol. 2020 Feb;27(3):269-271.
Here are excerpts:
The early detection of persons at risk remains the cornerstone of prevention. Heritability in common diseases is increasingly being recognized as relevant for cardiovascular practice, both by clinicians and by family members of those affected.
Nowadays, screening is advocated in families of patients with Bicuspid Aortic Valve disease (BAV). It is reported that first-degree relatives have a risk of between 6.4% and 9% to be affected, suggesting a number needed to screen of around 10 in BAV.1 It is, therefore, recommended in guidelines to have an echocardiographic screening of first-degree relatives.2 The question arises: should this likewise be recommended
for the other common valve anomaly, mitral valve prolapse (MVP)? The article by Hiemstra et al. in this issue3 adds evidence in this direction.MVP prevalence and pathophysiology
The finding of MVP in a patient with relatively non-specific symptoms such as atypical chest pain or palpitations is a frequent clinical scenario.4 With a prevalence of 1%, MVP is commonly encountered in an unselected community sample such as the Framingham study.5
[MVP results from development of] redundancy of valve tissue [which] may be local (‘‘fibroelastic deplete’’) or diffuse (‘‘Barlow phenotype’’) and disrupts mitral valve physiology in several characteristic ways, including shifting of coaptation points, valve regurgitation and elongation or rupture of the chordae tendineae, as well as dissociation of the mitral annulus and systolic ‘‘curling shape’’ dynamics of the basolateral left ventricular wall.6
Complications of MVP: regurgitation and ventricular arrhythmias
Undiagnosed and clinically silent MVP with a benign course is a common scenario.
However, an estimated subset of 7% of patients develop hemodynamically significant MVP-related regurgitation in which hypertension is a known accelerating factor/stressor on top of genetically susceptible valves and chordae.5
MVP has been linked to ECG changes, QT abnormalities and
late afterdepolarizations, with stretch and fibrosis of the papillary muscles contributing to ventricular arrhythmia on electrophysiological evaluations.A recent review on this topic by Basso et al.6 described
that the clinical challenge is the identification of individuals at risk for developing both hemodynamically relevant and arrhythmic MVP.In view of the above, mild – apparently insignificant – MVP may potentially represent not just an echocardiographic subtlety present within the community5 but rather a clinically
relevant entity requiring long-term hemodynamic and arrhythmic surveillance in a subset of patients.
The emerging role of family history and genetics
Various Mendelian inheritance patterns for MVP have
been recognized for decades.9Regarding the genetics of MVP, several gene variants, mainly in
the DCHS1 and FLNA genes, causing developmental errors in valve morphogenesis have been described.11,12Moreover, a Genome Wide Association Study identified six risk loci.13 Genetic MVP can also occur as part of a syndrome, like the connective tissue diseases Marfan syndrome and Ehlers Danlos syndrome.
While the knowledge of the familial basis for MVP has progressed, as of yet this is not routinely integrated in clinical practice. Previous large-scale work from the Framingham Study by Delling et al.5 demonstrated that the prevalence of MVP in the community is 1%, but the offspring of patients with
MVP have a significantly higher (5.4%) absolute risk of MVP.With this in mind, Hiemstra et al.3 investigated the heritability of MVP including a smaller (n ¼ 385) but more targeted subset of index patients with a more severe phenotype of mitral regurgitation referred for surgical repair. Family history data followed a systematic methodology including genetic counseling
and retrieval of relevant medical information in all patients suspected of familial MVP. They found that, in such a population, based on self-reported information of the index patients, familial occurrence is significant and ranges from 8% in relatives of patients with the fibroelastic-deplete phenotype to 26% in relatives of patients with Barlow phenotype. Of note, familial MVP was also found to correlate with a positive family
history for sudden cardiac death, which underscores the clinical relevance of the arrhythmic complications of MVP. Future studies to confirm these findings should consist of systematic screening of family members by cardiac imaging and obtaining sufficient follow-up data.Is ‘‘cascade’’ screening justified in more advanced MVP phenotypes?
A ‘‘surgical repair’’ or ‘‘referral type’’ population represents a more significant phenotype. With an absolute risk ranging between 8% and 26%, the numbers needed to screen range from 4 to 12.
However, several aspects need to be considered.
At what age should the screening of relatives start?
Is a single transthoracic echocardiography recording sufficient, or is there a role of repeated screening in case of negative result, and if so at what intervals?
Is there a role of genetic testing?
What is the surveillance policy in case of MVP, is there a role to screen for non-valvular symptoms such as arrhythmias, and at what intervals?
Figure 1 outlines a proposed algorithm.
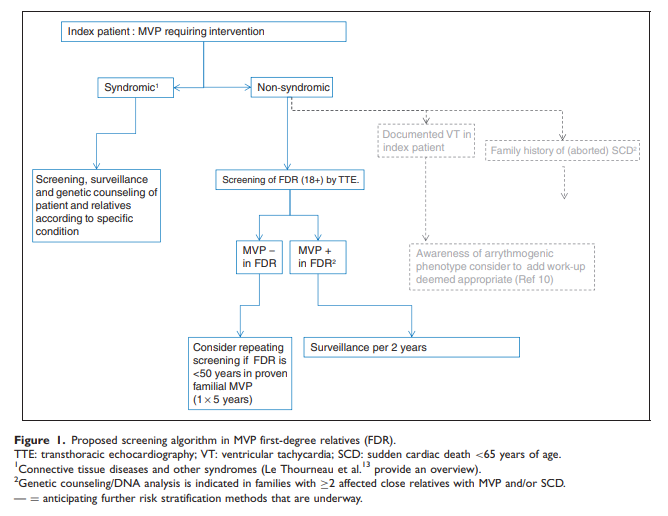
Lastly, it is important to consider that these findings are relevant for ‘‘non-syndromic’’ prolapse, and patients with a known associated syndrome and their relatives should receive surveillance and genetic counselling as appropriate.
In conclusion, the current contribution by Hiemstra et al.3 in this issue of EJPC raises awareness and provides evidence to advocate screening family members in such a referral-type
population of MVP patients. However, before this is implemented in clinical routine, several question remain to be addressed.



