
Evidence-based guideline summary: Diagnosis and treatment of limb-girdle and distal dystrophies: Report of the Guideline Development Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. [PubMed Abstract] [Full Text HTML] [Full Text PDF]. Neurology. 2014 Oct 14;83(16):1453-63.
The following post is just excerpts from the outstanding guideline (and be sure to visit the Muscular Dystrophy Association page on limb-girdle dystrophies):
Abstract
Objective:
To review the current evidence and make practice recommendations regarding the diagnosis and treatment of limb-girdle muscular dystrophies (LGMDs).
Methods:
Systematic review and practice recommendation development using the American Academy of Neurology guideline development process.
Results:
Most LGMDs are rare, with estimated prevalences ranging from 0.07 per 100,000 to 0.43 per 100,000. The frequency of some muscular dystrophies varies based on the ethnic background of the population studied. Some LGMD subtypes have distinguishing features, including pattern of muscle involvement, cardiac abnormalities, extramuscular involvement, and muscle biopsy findings. The few published therapeutic trials were not designed to establish clinical efficacy of any treatment.
Principal recommendations:
For patients with suspected muscular dystrophy, clinicians should use a clinical approach to guide genetic diagnosis based on clinical phenotype, inheritance pattern, and associated manifestations (Level B). Clinicians should refer newly diagnosed patients with an LGMD subtype and high risk of cardiac complications for cardiology evaluation even if they are asymptomatic from a cardiac standpoint (Level B). In patients with LGMD with a known high risk of respiratory failure, clinicians should obtain periodic pulmonary function testing (Level B). Clinicians should refer patients with muscular dystrophy to a clinic that has access to multiple specialties designed specifically to care for patients with neuromuscular disorders (Level B). Clinicians should not offer patients with LGMD gene therapy, myoblast transplantation, neutralizing antibody to myostatin, or growth hormone outside of a research study designed to determine efficacy and safety of the treatment (Level R). Detailed results and recommendations are available on the Neurology® Web site at Neurology.org.
—————————————————————————–
—————————————————————————–
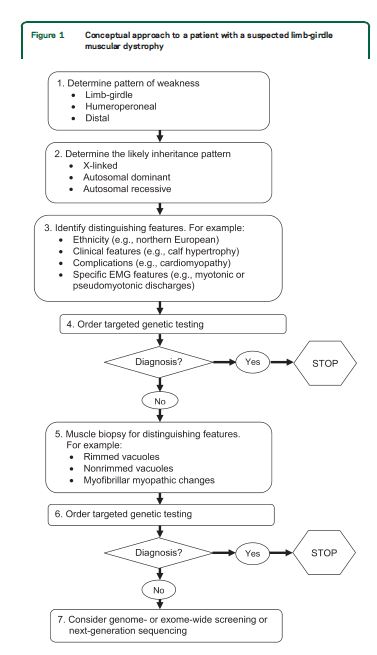
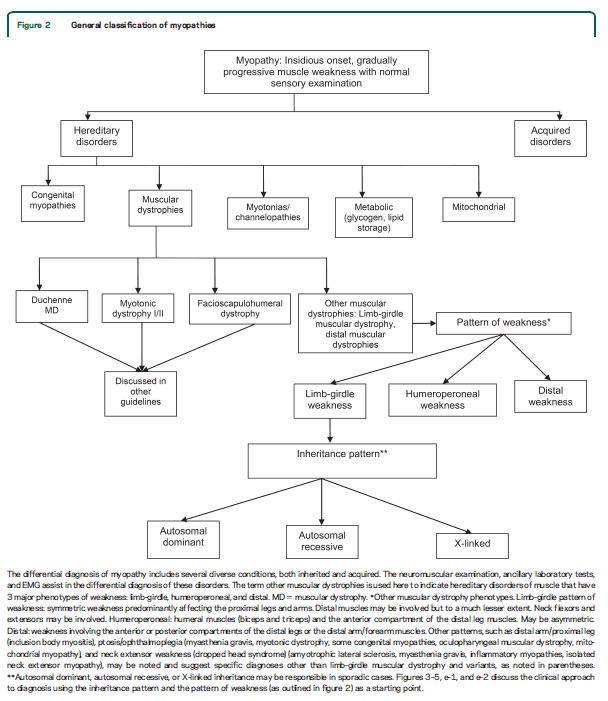
Limb-girdle muscular dystrophies (LGMDs) are a group of hereditary myopathies characterized by predominantly proximal muscle weakness (pelvic and shoulder girdles).1 Initially described as a clinical phenotype, they are now recognized as a heterogeneous group of myopathies that vary in severity and may affect persons at all ages from childhood through adulthood. The LGMDs are classified into 2 main groups depending on the inheritance pattern: LGMD1, autosomal dominant; and LGMD2, autosomal recessive. Appended to this numeric division is a letter designating the order of discovery for each chromosomal locus.2,3
This guideline reviews the current evidence and makes practice recommendations regarding the diagnosis and treatment of LGMDs. We also review other hereditary myopathies that may be considered forms of LGMD (e.g., hereditary inclusion body myopathies [hIBMs], Emery-Dreifuss muscular dystrophy [EDMD], Becker muscular dystrophy [BMD], and manifesting carriers of dystrophin mutations). We also discuss non–limb-girdle adult-onset myopathies that are genotypically identical to the LGMDs (e.g., Miyoshi distal myopathy, which is allelic to LGMD2B) and myofibrillar myopathies (MFM).
Duchenne dystrophy, congenital muscular dystrophy, myotonic dystrophy, and facioscapulohumeral dystrophy are not included in this guideline, as they will be discussed in forthcoming guidelines. Table e-1 on the Neurology® Web site at Neurology.org delineates the most recent classification of what is considered a muscular dystrophy in adults who were included in this review. We use the terms LGMD and muscular dystrophy interchangeably to refer to the disorders reviewed in this guideline.
The principal audience for this guideline is clinicians caring for patients with muscular dystrophies.
—————————————————————————–
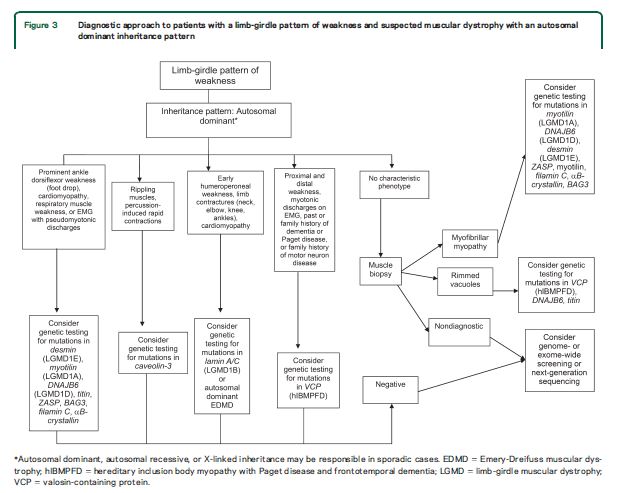
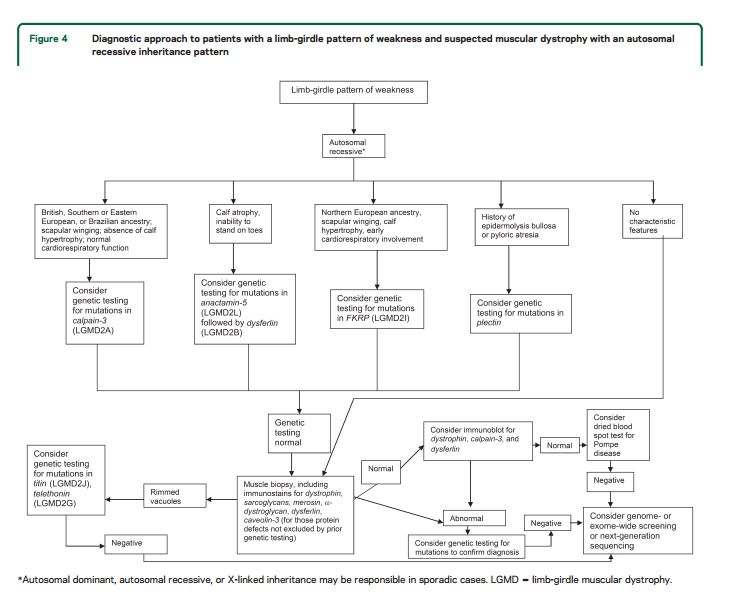
Our systematic review identified features common to most patients with muscular dystrophy. Most patients present with slowly progressive symmetrical weakness. The age at onset is usually adolescence to early adulthood but is highly variable, ranging from early childhood (LGMD2N, LGMD2P, LGMD1E, FHL1, BAG3)15,–40,e1,e2 to late adult life (e.g., Welander myopathy, Udd distal myopathy).e3–e11 Although the limb-girdle pattern of weakness affecting proximal muscles of the arms and legs is the most common, other patterns, including scapuloperoneal weakness and distal weakness, are not rare. Indeed, a single genotypic variety can present with different patterns of weakness in different patients. For example, mutations in the titin gene can manifest with limb-girdle weakness, a distal myopathy affecting predominantly anterior leg compartment muscles (Udd distal myopathy), or early ventilatory/respiratory failure.e3,e8–e17Patients with desmin mutations can present with limb-girdle, scapuloperoneal, or distal pattern of muscle weakness.29,32,34,36,–39,e1Lamin A/C mutations cause both limb-girdle and humeroperoneal phenotypes.e18–e23 Conversely, a single phenotype can result from more than one genotype. Miyoshi myopathy, manifesting with asymmetric leg weakness affecting posterior leg compartment muscles, can be seen in mutations indysferlin (LGMD2B)e24–e38 and ANO5 (LGMD2L).e39–e42 The humeroperoneal pattern of weakness is seen in EDMD, which can be due to mutations in the emerin,e19,e43,e44lamin A/C,e18–e23FHL1,20,22,e45TMEM43/LUMA,e46 and nesprin 1 and 2 genes (not reviewed in this guideline). Serum CK levels vary widely between patients with the same disorder, ranging from normal to greater than 10 times above normal levels, and can be as much as 100 times normal in some cases (e.g., LGMD2B).e36 EMG shows short-duration, small-amplitude motor units with early recruitment in weak muscles; findings may be subtle in mild cases. Routine muscle biopsy often shows nonspecific myopathic features, but some dystrophies have specific diagnostic features (discussed below).
Distinguishing features.
Although there are few pathognomonic features, many LGMD disorders have distinguishing features. These features for the major LGMD disorders are enumerated in table e-2. In addition to inheritance patterns and overall patterns of weakness, distinguishing clinical characteristics include the early development of foot drop (e.g., MFM27,30,32−34,36,38,39,e47–e65); asymmetry in muscle weakness (e.g., LGMD1A,27,e47–e54 LGMD2L,e39–e42,e66,e67 MFM 27,30,32−34,36,38,39,e47–e65); limb contractures (lamin A/C myopathies,e18–e23 EDMD,e43,e68–e70BAG325,e59); prominent muscle cramps (LGMD1Ce71–e73); family or personal history of frontotemporal dementia, Paget disease of bone, or motor neuron disease (hIBMPFD)e74–e90; ancestry (e.g., northern European for LGMD2Ie91); scapular winging (e.g., sarcoglycanopathies,12,13,e92–e94 LGMD2Ae95–e100); calf hypertrophy (e.g., BMD,10,11,e101–e152LGMD2Ie91,e153–e164); cardiomyopathy (e.g., LGMD2I,e91,e153,e154,e156,e160–e172BMDe101,e103,e104,e107,e108,e111,e113,e114,e116,e120–e122,e124,e126,e129,e132,e133,e137,e139,e151,e152); or cardiac conduction system abnormalities (e.g., laminopathy,e18,e173–e183 desminopathy27,29−40,e1,e2). Rippling muscle phenomenon and percussion-induced muscle contractions are noted in LGMD1C.e184,e185Epidermolysis bullosa or congenital pyloric atresia suggest plectin mutations.e186–e215 Distinguishing EMG features include myotonic and pseudomyotonic discharges (the latter characterized by runs of decrescendo positive sharp-wave discharges without the typical waxing and waning of amplitudes and frequencies) in MFM. Muscle biopsy features that can distinguish muscular dystrophies include the presence of rimmed vacuoles (e.g., LGMD1D, hIBM,e76,e77,e89,e216–e221 MFM), reducing bodies/cytoplasmic bodies (FHL119,e222–e225), and derangement of myofibrils consistent with MFM (desmin,e226,e227myotilin,27,e47,e48,e52 αB-crystallin,e49,e55–e57ZASP,27,e49,e58,e228 BAG3,25,26DNAJB6,e229filamin Ce49,e60–e62). Nemaline rods may be seen in distal myopathies due to nebulin mutations.e230,e231Reductions of specific proteins on immunohistochemistry suggest deficiencies of these proteins, although the diagnosis needs to be confirmed by genetic testing.
—————————————————————————–
Are there effective therapies for muscular dystrophies? The systematic review identified only 12 studies evaluating treatments for patients with LGMD. These [studies] are summarized [on 1455 + 1456 of the pdf.] [Short answer: not really–treatment should not be outside of a clinical trial–see below]
—————————————————————————–
—————————————————————————–
—————————————————————————–
—————————————————————————–
—————————————————————————–
MAJOR PRACTICE RECOMMENDATIONS
The recommendations below encompass 3 major areas: diagnosis, evaluation, and management of muscular dystrophies. The full recommendation set is available online.
Diagnosis of muscular dystrophies
The case for genetic diagnosis
Accurate diagnosis of the muscular dystrophies is important for patients, their families, and efficient and cost-effective use of medical resources
Diagnosis assists in defining the long-term prognosis, since some dystrophies are more rapidly progressive, involve the cardiorespiratory systems more frequently, or are associated with other disorders. The identification of these dystrophies through genetic testing will not only inform long-term prognosis but will also assist in directing care more efficiently (e.g., more frequent cardiorespiratory monitoring and prophylactic treatments such as pacer/defibrillator placement for those disorders known to be associated with cardiac involvement). Precise identification of the disorder also eliminates the need for repeated testing for an acquired, treatable disorder such as an inflammatory myopathy, because some dystrophies have inflammation on muscle biopsy, making diagnosis difficult on the basis of routine biopsy findings. In addition, the temptation to try immunosuppressive agents repeatedly, looking for a therapeutic response, is not unusual when there is no diagnosis and the patient is worsening. This exposes patients to potentially serious side effects of immunosuppressive medications. Patients on immunosuppressants need regular monitoring, adding logistical difficulties to a population that may have significantly impaired mobility. Health care costs are increased by repeated investigations, immunosuppressive treatments, and laboratory monitoring. Although establishing a genetic diagnosis is expensive on the front end, the costs of continued investigation for other causes and the risks and expenses associated with empiric trials of immunosuppressants make a strong case for establishing a genetic diagnosis, which often provides patients a sense of closure. Establishing a genetic diagnosis is crucial for genetic counseling to inform decision-making about having children and for screening of offspring. Treatment of cardiomyopathy, arrhythmias, and ventilatory failure prolongs life and improves quality of life in patients with other neuromuscular diseases.e242–e247
The approach to genetic diagnosis
Recommendations.
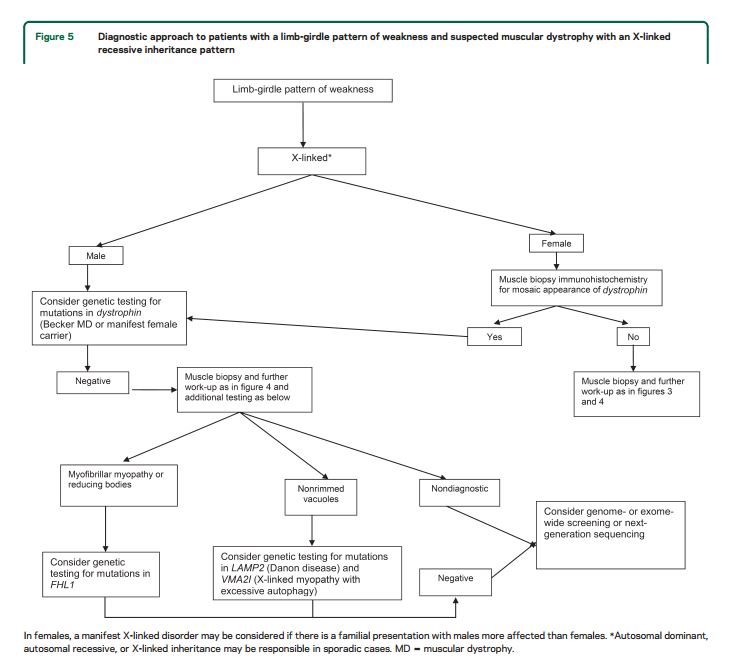
For patients with suspected muscular dystrophy, clinicians should use a clinical approach to guide genetic diagnosis based on the clinical phenotype, including the pattern of muscle involvement, inheritance pattern, age at onset, and associated manifestations (e.g., early contractures, cardiac or respiratory involvement) (Level B).
In patients with suspected muscular dystrophy in whom initial clinically directed genetic testing does not provide a diagnosis, clinicians may obtain genetic consultation or perform parallel sequencing of targeted exomes, whole-exome sequencing, whole-genome screening, or next-generation sequencing to identify the genetic abnormality (Level C).
Evaluation and medical management of muscular dystrophies.
Cardiac involvement
Recommendations
Clinicians should refer newly diagnosed patients with (1) LGMD1A, LGMD1B, LGMD1D, LGMD1E, LGMD2C–K, LGMD2M–P, BMD, EDMD, and MFM or (2) muscular dystrophy without a specific genetic diagnosis for cardiology evaluation, including ECG and structural evaluation (echocardiography or cardiac MRI), even if they are asymptomatic from a cardiac standpoint, to guide appropriate management (Level B).
If ECG or structural cardiac evaluation (e.g., echocardiography) has abnormal results, or if the patient has episodes of syncope, near-syncope, or palpitations, clinicians should order rhythm evaluation (e.g., Holter monitor or event monitor) to guide appropriate management (Level B).
Clinicians should refer muscular dystrophy patients with palpitations, symptomatic or asymptomatic tachycardia or arrhythmias, or signs and symptoms of cardiac failure for cardiology evaluation (Level B).
It is not obligatory for clinicians to refer patients with LGMD2A, LGMD2B, and LGMD2L for cardiac evaluation unless they develop overt cardiac signs or symptoms (Level B).
Dysphagia and nutrition
Recommendation
Clinicians should refer muscular dystrophy patients with dysphagia, frequent aspiration, or weight loss for swallowing evaluation or gastroenterology evaluation to assess and manage swallowing function and aspiration risk, to teach patients techniques for safe and effective swallowing (e.g., chin tuck maneuver, altered food consistencies), and to consider placement of a gastrostomy/jejunostomy tube for nutritional support (Level B).
Pulmonary complications
. . . . Patients with respiratory failure from neuromuscular-related weakness often do not have symptoms, such as dyspnea, that precede the onset of respiratory failure. Impending respiratory failure in these patients is often identified only with pulmonary function tests. Respiratory failure constitutes a major source of morbidity, interfering with daytime cognitive function and negatively affecting quality of life. In addition, ventilatory and oropharyngeal weakness can threaten survival through the risk of upper airway obstruction or bellows failure (or both).e243 Patients with respiratory failure secondary to muscle weakness often have improved quality of life with noninvasive pulmonary ventilation.e243
Recommendations
Clinicians should order pulmonary function testing (spirometry and maximal inspiratory/expiratory force in the upright and, if normal, supine positions) or refer for pulmonary evaluation (to identify and treat respiratory insufficiency) in muscular dystrophy patients at the time of diagnosis, or if they develop pulmonary symptoms later in their course (Level B).
In patients with a known high risk of respiratory failure (e.g., those with LGMD2I or MFM), clinicians should obtain periodic pulmonary function testing (spirometry and maximal inspiratory/expiratory force in the upright position and, if normal, in the supine position) or evaluation by a pulmonologist to identify and treat respiratory insufficiency (Level B).
It is not obligatory for clinicians to refer patients with LGMD2B and LGMD2L for pulmonary evaluation unless they are symptomatic (Level C).
Clinicians should refer muscular dystrophy patients with excessive daytime somnolence, nonrestorative sleep (e.g., frequent nocturnal arousals, morning headaches, excessive daytime fatigue), or respiratory insufficiency based on pulmonary function tests for pulmonary or sleep medicine consultation for consideration of noninvasive ventilation to improve quality of life (Level B).
Spinal deformities
Recommendations
Clinicians should monitor patients with muscular dystrophy for the development of spinal deformities to prevent resultant complications and preserve function (Level B).
Clinicians should refer muscular dystrophy patients with musculoskeletal spine deformities to an orthopedic spine surgeon for monitoring and surgical intervention if it is deemed necessary in order to maintain normal posture, assist mobility, maintain cardiopulmonary function, and optimize quality of life (Level B).
Rehabilitative management and treatment of muscular dystrophies
Recommendations
Clinicians should refer patients with muscular dystrophy to a clinic that has access to multiple specialties (e.g., physical therapy, occupational therapy, respiratory therapy, speech and swallowing therapy, cardiology, pulmonology, orthopedics, and genetics) designed specifically to care for patients with muscular dystrophy and other neuromuscular disorders in order to provide efficient and effective long-term care (Level B).
Clinicians should recommend that patients with muscular dystrophy have periodic assessments by a physical and occupational therapist for symptomatic and preventive screening (Level B).
While respecting and protecting patient autonomy, clinicians should proactively anticipate and facilitate patient and family decision-making as the disease progresses, including decisions regarding loss of mobility, need for assistance with activities of daily living, medical complications, and end-of-life care (Level B).
For patients with muscular dystrophy, clinicians should prescribe physical and occupational therapy, as well as bracing and assistive devices that are adapted specifically to the patient’s deficiencies and contractures, in order to preserve mobility and function and prevent contractures (Level B).
Strength training and aerobic exercise training
Recommendations
Clinicians may advise patients with muscular dystrophy that aerobic exercise combined with a supervised submaximal strength training program is probably safe (Level C).
Clinicians may advise patients with muscular dystrophy that gentle, low-impact aerobic exercise (swimming, stationary bicycling) improves cardiovascular performance, increases muscle efficiency, and lessens fatigue (Level C).
Clinicians may counsel patients with muscular dystrophy to hydrate adequately, not to exercise to exhaustion, and to avoid supramaximal, high-intensity exercise (Level C).
Clinicians should educate patients with muscular dystrophy who are participating in an exercise program about the warning signs of overwork weakness and myoglobinuria, which include feeling weaker rather than stronger within 30 minutes after exercise, excessive muscle soreness 24–48 hours following exercise, severe muscle cramping, heaviness in the extremities, and prolonged shortness of breath (Level B).
Medical treatments
Recommendation
Clinicians should not currently offer patients with muscular dystrophy gene therapy, myoblast transplantation, neutralizing antibody to myostatin, or growth hormone outside of a research study designed to determine the efficacy and safety of the treatment (Level R).



