
This post consists of excerpts from Resource (1) below, How I Treat Unexplained Refractory Iron Deficiency Anemia [PubMed Abstract] [Full Text HTML] [Full Text PDF]. Blood. 2014 Jan 16;123(3):326-33. doi: 10.1182/blood-2013-10-512624. Epub 2013 Nov 8:
Abstract:
Endoscopic gastrointestinal workup fails to establish the cause of iron deficiency anemia (IDA) in a substantial proportion of
patients. In patients referred for hematologic evaluation with unexplained or refractory IDA, screening for celiac disease,
autoimmune gastritis, Helicobacter pylori, and hereditary forms of IDA is recommended. About 4% to 6% of patients with
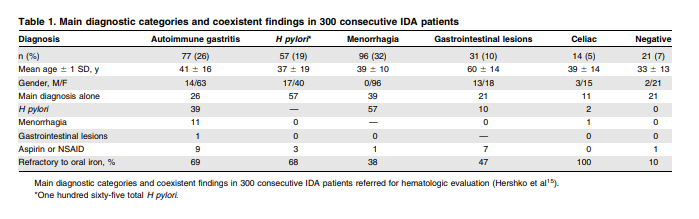
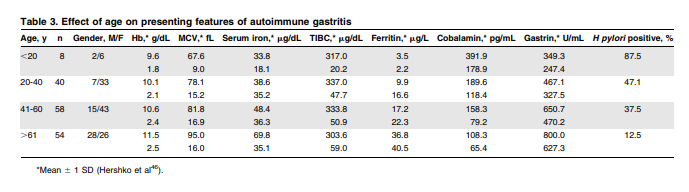
obscure refractory IDA have celiac disease, and autoimmune gastritis is encountered in 20% to 27% of patients. Stratification by age cohorts in autoimmune gastritis implies a disease presenting as IDA many years before the establishment of clinical cobalamin deficiency. Over 50% of patients with unexplained refractory IDA have active H pylori infection and, after excluding all other causes of IDA, 64% to 75% of
such patients are permanently cured by H pylori eradication. In young patients with a history suggestive of hereditary
iron deficiency with serum ferritin higher than expected for IDA, mutations involving iron trafficking and regulation should be
considered. Recognition of the respective roles of H pylori, autoimmune gastritis, celiac disease, and genetic defects in the
pathogenesis of iron deficiency should have a strong impact on the current diagnostic workup and management of
unexplained, or refractory, IDA. (Blood. 2014;123(3):326-333)Introduction:
In the present article, refractoriness to oral iron is defined as
failure to respond to treatment at a dose of at least 100 mg of
elemental iron per day after 4 to 6 weeks of therapy. Subjects in
whom IDA is unresponsive to standard oral iron treatment or in
whom anemia persists despite a negative gastrointestinal workup represent an important subgroup of patients referred for hematologic evaluation.Studies in the late 90s have established celiac disease as
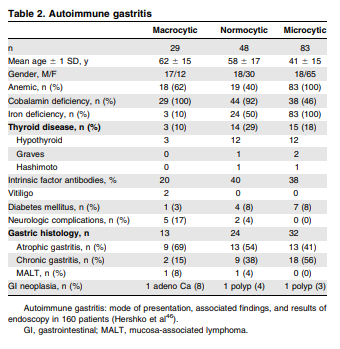
a possible cause of IDA refractory to oral iron treatment, without other apparent manifestations of malabsorption syndrome.10 In addition, Helicobacter pylori has been implicated in several earlier studies as a cause of IDA refractory to oral iron treatment, with a favorable response to H pylori eradication.11,12 Likewise, autoimmune atrophic gastritis, a condition associated with chronic idiopathic iron deficiency, has been shown to be responsible for refractory IDA in over 20% of patients with no evidence of gastrointestinal blood loss.13,14The availability of convenient, noninvasive screening methods
for identifying celiac disease (anti-tissue transglutaminase
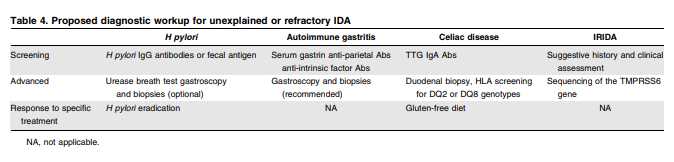
[TTG] antibodies), autoimmune atrophic gastritis (serum gastrin, parietal cell, or intrinsic factor antibodies), and H pylori infection (antibody screening or fecal antigen and urease breath test), and the recent recognition of rare inherited forms of iron deficiency greatly facilitated the diagnosis of these entities, resulting in an increased awareness of these conditions and their possible role in the causation of IDA.
Hereditary microcytic anemias
A discussion of unexplained refractory IDA would be incomplete
without mention of the rare hereditary forms of IDA. These novel forms of microcytic anemia have been identified following the remarkable recent progress made in the understanding of molecular mechanisms involved in iron trafficking and regulation and include defects in iron absorption, transport, utilization, and recycling.75 Among these rare microcytic anemias, iron refractory IDA (IRIDA) is considered as the
most ”frequent” 76 and will be discussed in some detail.Hereditary iron-refractory iron deficiency syndrome (IRIDA)
IRIDA is an autosomal-recessive disorder caused by mutations on the transmembrane serine protease 6 (TMPRSS6) gene encoding Matriptase-2 (MT-2).77 MT-2 is a transmembrane serine protease essential for downregulating hepcidin, the master regulator of iron homeostasis. In vitro studies in transfected hepatic cell lines suggest that MT-2 cleaves hemojuvelin, an activator of hepcidin expression.78 This function is impaired in mutations involving TMPRSS6 resulting in IRIDA. Hence, serum hepcidin is inappropriately high and patients are unable to respond to iron deprivation by suppressing hepcidin expression.79-81
Patients with hereditary IRIDA have hypochromic microcytic
anemia with very low serum iron and transferrin saturation. However, unlike nutritional iron deficiency, their serum ferritin is normal or inappropriately high for the degree of iron deficiency. The severity of anemia is variable, usually moderate; it is not present at birth and develops after the neonatal period. Although the anemia tends to be more severe in childhood than later, there is no correlation between age at diagnosis and the severity of anemiaDiagnosis of IRIDA requires identification of genetic lesions by
sequencing the exons and exon-intron boundaries of the TMPRSS6 gene.83,84 Causal mutations are nucleotide changes that interfere with the protein synthesis or amino acid substitutions that are spread over all the extracellular domains of the protein. Although some mutants fully suppress the proteolytic activity, others maintain a residual activity in in vitro studies using the hepcidin promoter.85 Up to the present, over 50 patients from 34 families have been reported with a wide range of ethnic origins representing .40 different TMPRSS6 mutations.76,86,87As implied by the term IRIDA, patients are unable to respond
satisfactorily to oral iron treatment.ACD (Anemia of Chronic Disease) may resemble IRIDA in many respects, as proinflammatory cytokines stimulate hepcidin synthesis and are associated with anemia, low serum iron, and increased serum ferritin. Differential diagnosis with IRIDA is based on the mild normocytic or moderately microcytic nature of the anemia in ACD, combined with clinical and laboratory evidence of inflammation.
Diagnostic workup and treatment of unexplained refractory IDA
Diagnosis
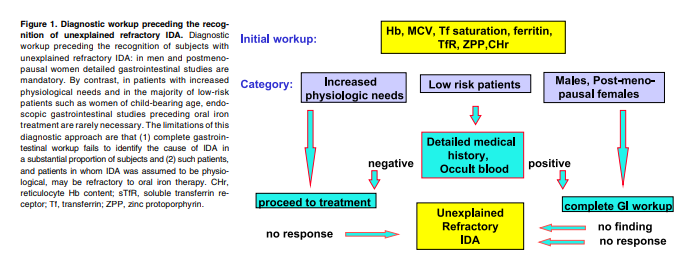
As stated earlier, our definition of refractory anemia is failure to respond to oral iron treatment with 100 mg of elemental iron per day for 4 to 6 weeks by an increase in Hb of at least 1 g/dL. Other causes of treatment failure should be excluded first. Admittedly, this is not an easy task as even experienced clinicians may be misled by low compliance, inaccurate history, false diagnosis, and occasional factitious anemia. Care should be taken to exclude ACD using C-reactive protein and other acute-phase reacting proteins, chronic renal failure, use of proton pump inhibitors impairing gastric acidity, and gastrointestinal bleeding induced by drugs or by hereditary bleeding disorders.
Patients with positive H pylori serology or fecal antigen should
be referred for urease breath test to confirm H pylori infection.
Endoscopic confirmation of H pylori gastritis is not mandatory.
Patients with autoimmune gastritis as indicated by the combined presence of increased serum gastrin and parietal cell or intrinsic factor antibodies should be screened for thyroid disease and for cobalamin depletion. Upper gastrointestinal endoscopy with mucosal biopsy is recommended for all new autoimmune gastritis patients at diagnosis and thereafter at 5- to 10-year intervals. Patients with positive celiac serology should be referred for duodenal biopsy and tested for the
HLA-DQ2 and -DQ8 genotypes.In patients with a history suggestive of hereditary iron deficiency with serum ferritin higher than expected, a genetic workup is recommended.75 For suspected IRIDA, the National Center for Biotechnology Information (NCBI) site of available laboratories for TMPRSS6 sequencing is listed in the Appendix.
Intravenous iron therapy
Intravenous iron replacement is indicated for patients failing to
respond to oral iron treatment. Several IV iron preparations are
now available.101 They consist of a core of iron oxohydroxide with a protective carbohydrate shell of sugar polymers.102 Following injection, iron is slowly released and is picked up by plasma transferrin. The rate of dissociation of iron from the complex is variable. Iron gluconate is the most labile with the most rapid release of iron and, consequently, doses of 62.5 to 125 mg are the highest that can be tolerated at a single infusion. Iron sucrose is more stable and higher doses, usually 200 mg of iron, may be given safely. The most stable iron preparations are the iron dextrans tightly bound to their iron oxohydroxide cores. There are highmolecular-weight and low-molecular-weight iron dextrans. According to recent pharmacovigilance reports from North America and Europe, the OR of reported total absolute rates of adverse life-threatening events with parenteral iron is 38 per million doses: the risk of anaphylaxis with iron dextran compared with iron sucrose is 17.7- to 16.9-fold.103 In view of the increased risk of life-threatening adverse effects and the availability of much safer IV iron compounds, the use of high-molecular iron dextran is no longer justified.Conclusions
In high-risk IDA patients, complete endoscopic workup is mandatory. By contrast, low-risk patients, representing the vast majority of subjects with refractory IDA, remain the responsibility of the hematologist. The hematologist, in turn, is now equipped with powerful noninvasive tools to identify IDA patients requiring special attention. Recognition of the respective roles of H pylori, autoimmune gastritis, celiac disease, and inherited disease in the pathogenesis of iron deficiency should have a strong impact on the current diagnostic workup and management of refractory IDA and a sharp decrease in the proportion of IDA patients still labeled “unexplained.”
Resources:
(1) How I Treat Unexplained Refractory Iron Deficiency Anemia [PubMed Abstract] [Full Text HTML] [Full Text PDF]. Blood. 2014 Jan 16;123(3):326-33. doi: 10.1182/blood-2013-10-512624. Epub 2013 Nov 8



