
Children and adults who are on a ketogenic diet require careful and thorough evaluation when they exhibit acute illness. Please see clinical guideline Ketogenic diet acute illness management from The Royal Children’s Hospital Melbourne. Although the guideline is for children, the same management principles would apply for adults.
This post consists of excerpts from the 2018 Guidelines [Resource (1)] Optimal clinical management of children receiving dietary therapies for epilepsy: Updated recommendations of the International Ketogenic Diet Study Group [29881797]:
Key Points
• This manuscript represents a re-evaluation of ketogenic
diet best practices, 10 years after the previous
guideline publication
• Ketogenic diets should be used in children after 2 antiseizure
drugs have failed, and for several epilepsy syndromes
perhaps even earlier
• There are 4 major ketogenic diets, and the one chosen
should be individualized based on the family and child
situation
• Flexibility in the initiation of ketogenic diets is appropriate,
with fasting and inpatient initiation optional
• Children should be seen regularly by the ketogenic
diet team, along with labs and side effect monitoring
at each visit.Ketogenic dietary therapies (KDTs) are well-established,
nonpharmacologic treatments used for children and adults
with medication-refractory epilepsy. . . . There are currently 4
major KDTs: the classic KD, the modified Atkins diet
(MAD), the medium chain triglyceride diet (MCT), and the
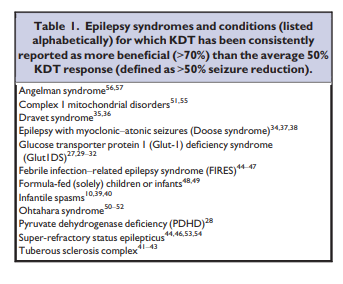
low glycemic index treatment (LGIT).There are several specific conditions for which the group
considered that KDTs should be used early in the course of
epilepsy management (Table 1).
Preliminary experience also showing some beneficial
effects of the KD has been reported in epileptic encephalopathies
such as Lafora body disease,58 Rett syndrome,59,60
Landau-Kleffner syndrome,61 and subacute sclerosing
panencephalitis (Table 2).62 Single reports describe the use
of the diet in metabolic disorders such as phosphofructokinase
deficiency, adenylosuccinate lyase deficiency, and
glycogenosis type V.63–65 Other conditions with limited
data include juvenile myoclonic epilepsy,66 CDKL5
encephalopathy,67 epilepsy of infancy with migrating focal
seizures,68 childhood absence epilepsy,69 and epileptic
encephalopathy with continuous spike-and-wave during
sleep.70
All of the above strong and moderate indications have
sufficient data in our guideline group’s opinion to justify the
use of KDT. However, are these reports enough to justify
early or even first-line use? Many of these conditions have
comorbid cognitive or behavioral abnormalities; would
KDT ameliorate these issues as well? In addition, several of
these indications have “preferred” ASDs (eg, valproate and
clobazam for Dravet syndrome, corticosteroids and vigabatrin
for infantile spasms). Does KDT work synergistically
with these ASDs for these particular conditions? These
important questions should be investigated in the years to
come.KDTs are contraindicated in several specific disorders
(Table 3). The metabolic adaptation to KDT involves a shift
from use of carbohydrates to lipids as the primary energy
source. As such, a patient with a disorder of fat metabolism
might develop a severe deterioration in the setting of fasting
or KDTs. Therefore, before initiating the KDT, a child
should be screened for disorders of fatty acid transport and
oxidation if there is clinical concern for one of these conditions,
especially in the setting of epilepsy without a clear
etiology.
Before starting KDT, one should consider inborn errors of metabolism that could lead to a severe metabolic crisis and ruled out if there is a clinical suspicion for these disorders.
Pre-diet evaluation and counseling
A clinic visit prior to initiation of KDT is strongly
advised. The goals of this visit are to identify the seizure
type(s), rule out metabolic disorders that are contraindications
to KDT, and evaluate for complicating comorbidities (presence of kidney stones, swallowing difficulty, hypercholesterolemia, poor weight gain or oral intake, gastroesophageal reflux, constipation, cardiomyopathy, and chronic metabolic acidosis [Table 4]). Neurologists should review all current medications in partnership with a pharmacy and/or internet-based guides to determine carbohydrate content and options of switching to lower carbohydrate preparations while the patient is on KDT. [See Resource (2) below]Screening laboratory studies should be obtained prior to
starting KDT (Table 4).
Renal ultrasounds are advised only if there is a family history of kidney stones. A comprehensive evaluation should be undertaken if no clear etiology for the patient’s epilepsy has been identified. Electroencephalography and brain magnetic resonance imaging (MRI) are essential to identifying those patients who are possible surgical candidates.
A key component of KDT is the information the family
receives prior to the initiation of the diet. Helpful resources
for families include publications, websites, and videos from
support groups such as The Charlie Foundation [For Ketogenic Therapies] and Matthew’s Friends [Ketogenic Dietary Therapies] .6–8The expected length of time on KDT should be discussed [with the family]. A minimum of 3.2 months (SD 1.3) to allow for potential improvement to occur is advised.
Short-term difficulties are not uncommon at the time of KDT initiation and should be discussed with families; those occurring during the first week of the KD do not portend poor efficacy later.79 A social worker or nurse on the team can be instrumental in helping the family transition to KDT by assessing family needs, financial limitations, and gathering
resources, and contacting other families on KDT for parent-to-parent support.Specific diet selection and provision
There are 4 major KDTs with published evidence supporting
their use. The first 2 created, the classic long-chain
triglyceride (LCT) KD and the medium-chain triglyceride
(MCT) diet, have been in existence the longest and are typically
started in the hospital by a dietitian and neurologist. The LCT diet has been the more traditional KD treatment, for which most data are available, and is used by every center in this consensus group. The MCT diet may be preferable in some cases.81–84 In the classic KD, the fat source is predominantly LCT, which is obtained primarily from standard foods. MCT oils yield more ketones per kilocalorie of energy than LCTs. This increased ketogenic potential means less total fat is needed in the MCT diet, thus allowing the inclusion of more carbohydrate and protein and potentially food choices. There is no evidence for different efficacy between the MCT and classic KD (Class III
evidence).82,83The classic KD is calculated in a ratio of grams of fat to
grams of protein plus carbohydrate combined. The most
common ratio is 4 g of fat to 1 g of protein plus carbohydrate
(described as “4:1”); 90% of calories are from fat. A 3:1 or lower ratio can be used alternatively to increase protein or carbohydrate intake; this is more appropriate as well for diet initiation in infants. There is one publication reporting that a 4:1 ratio, when used at initiation, may be more advantageous for the first 3 months in older children, after which the ratio can be reduced.85The traditional MCT diet is used by 10 (40%) of the consensus
ketogenic diet centers and comprises 60% energy from MCT. This level of MCT can cause gastrointestinal discomfort in some children. To improve this difficulty, a modified MCT diet was created, using 30% energy from MCT oil, with an additional 30% energy from long chain fats.82 Many children will be on a 40–50% energy MCT diet, and can tolerate as high as 60% if necessary. MCT oil has also been used as a supplement to the classic KD to boost ketosis, improve lipid abnormalities, and due to laxative properties. MCT can be given in the diet as coconut oil or as an emulsion. MCT should be included in all meals
when used. Better toleration may be achieved using lessMCT with each meal but providing more meals per day. MCT oil consumption may also cause throat irritation due to the presence of C6 (caproic acid).88In the past 16 years, 2 other dietary therapies have been
developed for the treatment of epilepsy: the Modified Atkins Diet (MAD) and Low Glycemic Index Treatment (LGIT).24,25,89,90 Both of these KDTs are initiated universally as outpatients and they do not require precise weighing of food ingredients. They tend to require less dietitian time for meal calculations and allow more parental independence. The MAD is offered by 23 (92%) of consensus ketogenic centers and the LGIT by 17 (68%).The MAD is a high-fat, low-carbohydrate therapy similar
to the classic KD in food choices, and typically provides
approximately a 1:1–1.5:1 ketogenic ratio, but no set ratio is
mandated and some children can achieve as high as a 4:1
ratio.24 The initial daily carbohydrate consumption on the
MAD is approximately 10–15 g (comparable to the strict
initiation phase of the Atkins diet used for weight loss), with
a possible increase to 20 g per day after 1–3 months.90
However, there is no limitation on protein, fluids, or calories,
making meal planning easier. In addition, as detailed
calculations are not required, this diet may be ideal for low resource settings with a paucity of trained dietitians.The MAD has been shown to be effective in children with
refractory epilepsy in a randomized controlled trial (Class
III evidence). . . . For adolescents and adults, the MAD or LGIT is preferred by most (72%) of the consensus group largely due to better adherence.The LGIT was designed based on the hypothesis that
stable glucose levels play a role in the mechanism of the
KD.25 The LGIT allows liberalization of total daily carbohydrate intake to approximately 40–60 g/day, but favors
carbohydrates with low glycemic indices <50. A few uncontrolled studies suggest that the LGIT may be efficacious in
children with refractory epilepsy, but there are no trials comparing the LGIT with other KDTs.94–97 The LGIT has
been reported as particularly effective in children with Angelman syndrome, including a single center case series
of 23 patients.56Committee conclusions
The specific KDT chosen should be individualized based
on the family and child situation, rather than perceived efficacy,
together with the expertise of the KDT center. Calorie
and fluid restriction are no longer recommended. Children
younger than 2 years of age should be started on the classic
KD (Class III evidence), and a formula-based KD may be
helpful for this age group. There is reasonable evidence for
the use of the MCT (Class III), MAD (Class III), and LGIT,
and most consensus ketogenic centers are offering these options for KDT. These latter 2 therapies are recommended for adolescents, but centers may choose the classic KD for individual cases, especially in those with enteral feeding. The MAD is being studied for use in areas with limited resources.Supplementation
Due to the limited quantities of fruits, vegetables,
enriched grains, and foods containing calcium in KDT,
supplementation is essential, especially for B vitamins
(Table 5).
Committee Conclusions
All children should receive a daily multivitamin and calcium
with adequate vitamin D. Oral citrates appear to prevent
kidney stones (Class III); however, there was a mixed
opinion on empiric use. Vitamin D levels decrease on the
KD, but again, there were split opinions on empiric extra
supplementation. There is no recommendation for the
empiric use of antacids, laxatives, probiotics, exogenous
ketones, additional selenium, or carnitine with the KD at
this time.Maintenance of children receiving dietary therapies
Maintenance of children receiving dietary therapies
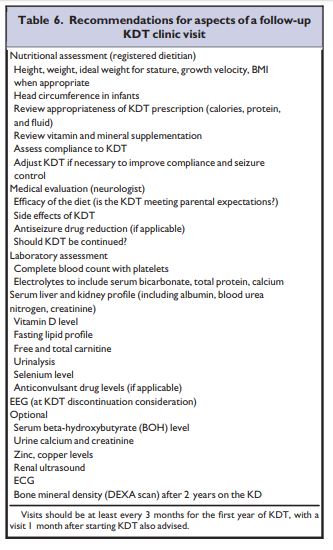
The child on KDT should be seen regularly for follow-up
evaluation by both dietitians and neurologists familiar with
KDT (Table 6).131,132
[Monitoring Of Ketones]
Urine ketones should be checked at home by parents several
times per week, preferably at different times of the day,
as urine ketones can be low in the morning and higher in the
evening. There are data regarding the value of serum BOH
(beta-hydroxybutyrate), with some studies suggesting that
serum BOH may better correlate with seizure control.134–136
Serum ketosis is more accurate, but more expensive and
requires finger sticks. The current guideline for infants on
the KD recommends serum BOH monitoring.14 Several
members of the consensus group suggested that obtaining
serum BOH at routine KDT clinic visits was valuable, especially
during the diet-initiation period, and 44% (11/25) of
the group have parents use home BOH meters. This was
higher than the previous consensus statement (15% recommending
home BOH meters). BOH monitoring may be reasonable
when urine ketones results do not clinically match
seizure control or fluctuate without explanation.In addition to a complete examination with accurate
weight and height measurement at each follow-up visit,
laboratory studies are recommended (Table 6 above).Nutritional ketosis can be adjusted through diet manipulation
to try to improve seizure control. Increasing the
fat content of the diet and lowering the carbohydrate
and/or protein is a method that can be trialled to induce
deeper ketosis. This is known as “increasing the ratio” in
classic KD terminology. Similarly, lowering the fat content
can reduce ketosis which may be desirable when
ketosis is too strong (ie, causing lethargy) or if weaning
KDT. Ketogenic practitioners have occasionally incorporated
MCT oil into the classic KD to achieve higher
ketosis while maintaining the same ratio and for its
added benefit as a laxative effect and easier digestibility
over long-chain fats.Adverse effects
Side effects of KDT occur, and neurologists and dietitians
need to understand how to manage them.129,142,143 The most
common involve the gastrointestinal system and are often
seen during the initial few weeks of dietary therapy. Constipation, emesis, and abdominal pain may occur in up to 50% of children.79,129,144 These symptoms are usually mild and easy to correct with minimal interventions. When adequately managed and prevented, gastrointestinal side effects
are rarely a reason to discontinue KDT.Hyperlipidemia is a well-known side effect of almost all
KDT.137,145–148 Increased serum triglycerides and total and
low-density lipoprotein (LDL) cholesterol levels have been
reported in 14–59% of children on the classic KD.129,137,147,148 Hyperlipidemia can be seen as early as the first month of therapy.148 Preliminary data suggest that despite an early increase in serum lipids during the first months of KDT, this increase is usually temporary. In one study, 60% of those on the classic KD had hypercholesterolemia (>200 mg/dl).147 By 12 months, the serum lipid values often normalize and remain within normal limits.137,149Although the risk for coronary artery disease may
increase with long-term elevations of cholesterol levels,
previous pediatric studies showed no change in the carotid
intima-media thickness compared to baseline at 6 and
12 months of therapy.150,151 On the other hand, local and
systemic arterial stiffness was significantly increased in 2
studies involving children and young adults, correlated with
serum cholesterol and triglycerides levels.152,153 Nevertheless,
long-term vascular outcomes of this high fat diet are
not known.Renal calculi historically have occurred in 3–7% of children
on KDT.115,127,154 They typically do not require KDT
discontinuation and lithotripsy is necessary only rarely.There is mixed data on the effect of KDT on growth in
children. However, all six studies with longer than 6 months
duration indicate that the classic KD has negative effects on
growth, and over time may cause a height deceleration.84,138,139,155–157Cardiac abnormalities have been reported in children on
the KD, including cardiomyopathy and prolonged QT interval.124,125,158,159 The mechanism of these complications is not fully understood; one case was associated with selenium
deficiency, but others were not. As stated previously, routine
ECG is not recommended at this time as a screening
test. Pancreatitis has also been reported.129,160 Hepatic dysfunction may be more likely to occur in children who are on
both valproic acid and KDT, with intercurrent viral illness
furthering increasing the risk of elevated transaminases.112,129,142The long-term complications in children maintained on
KDT for >2 years have not been reviewed systematically;
there is only one report in the literature looking at this small
subgroup.149 In this population, there was a higher risk of bone fractures, kidney stones, and decreased growth, but
dyslipidemia was not identified.149Committee Conclusion
Like all medical therapies KDTs have potential adverse
effects. Overall, the risk of serious adverse events is low;
KDTs do not need to be discontinued for most adverse
effects. Gastrointestinal complaints are often the most common
but can be mostly remedied.Discontinuation
A large percentage, 80%, of children who are seizure-free
on KDT will remain that way after KDT is discontinued.164
The risk may be higher in those with discharges on EEG,
brain malformations, and tuberous sclerosis complex.164 In
a multicenter study from Argentina, seizures recurred in
25% of those who were seizure-free and stopped the KD,
with a median period of follow-up after discontinuation of
the KD of 6 years.144 Many of these children with recurrent
seizures had brain lesions and EEG abnormalities.144



Resources:
(1) Optimal clinical management of children receiving dietary therapies for epilepsy: Updated recommendations of the International Ketogenic Diet Study Group [Pubmed Abstract] [Full Text HTML] [Full Text PDF]. Epilepsia Open. 2018 May 21;3(2):175-192. doi: 10.1002/epi4.12225. eCollection 2018 Jun.
(2) Medication Management On The Ketogenic Diet [Link is to the PDF]. By Jeff Curless, Pharm.D. Clinical Pharmacy Resident CHOC Children’s Hospital Email: jcurless@choc.org. Medications (pills) can have a lot of carbs in them. Dr. Curless explains how to deal with this problem.
(3) Ketogenic diet acute illness management, Clinical Guidelines (Nursing) from the Royal Children’s Hospital Of Melbourne. Updated November 2015.



